Lung cancer is the leading cause of cancer-related deaths globally. Despite current treatment approaches that include surgery, chemotherapy, radiation and immunotherapies, lung cancer accounted for 1.79 million deaths worldwide in 2020, emphasizing the urgent need to find novel agents and approaches for more effective treatment. Traditionally, chemicals derived from plants, such as paclitaxel and docetaxel, have been used in cancer treatment, and in recent years, research has focused on finding other plant-derived chemicals that can be used in the fight against lung cancer. Ursolic acid is a polyphenol found in high concentrations in cranberries and other fruits and has been demonstrated to have anti-inflammatory, antioxidant and anticancer properties. Here, researchers summarize studies examining the effects of ursolic acid and its derivatives on lung cancer. Data from in vitro cell culture and in vivo animal studies show potent anticancer effects of ursolic acid and indicate the need for clinical studies.
- lung cancer
- ursolic acid
- proliferation
- survival
- invasion
- metastasis
- signaling cascades
1. Effects of Ursolic Acid against Lung Cancer: In Vitro Studies
| Cell Type | Dose/Duration | Findings | Mechanism | Reference |
|---|---|---|---|---|
| A549 | UA 2–40 µM 0–72 h |
↓ Proliferation ↑ Apoptosis G1 phase cell cycle arrest |
↑ p53 protein ↓ Cyclin D1, D2 and E ↑ Fas/APO-1 receptor ↑ FasL ↑ Bax protein ↓ NF-kB/p65 activity ↓ Bcl-2 protein ↓ Bcl-Xl protein |
[1] |

2. Effects of Ursolic Acid against Lung Cancer: In Vivo Studies
| Xenograft Model | Dose/Duration | Findings | Mechanism | Reference | |
|---|---|---|---|---|---|
| 6–8-week nude mice A549 cells injected subcutaneously |
UA—10 mg/kg intragastrical administration/ 1 week |
↓ Tumor volume | Not investigated | [19] | |
| H460 | UA 3, 10 and 30 µM 24 h |
↑ Apoptosis ↓ Proliferation ↓ Migration |
↑ Cleaved caspase-3 ↑ MMP 1, 2, 3, 9 and 10 gene expression | ||
| C57 BL/6 mice injected with LLC-luciferase (1 × 107 cells/mouse) | UA—100 mg/kg intraperitoneally injected | ↑ Cytosolic glucocorticoid | receptor |
↓ Tumor volume ↓ Tumor weight[ |
↓ VRK1 activity4] |
| [ | 8 | ] | A549 H3255 Calu-6 |
UA 2, 4, 8 and 16 µM |
|
| Female Balb/c nude mice A549 cells 2 × 106 cells/mouse |
UA 50 or 100 mg/kg subcutaneous injection/every other day for 2 weeks |
↓ Tumor growth ↓ Tumor weight |
↓ MMP-2 ↓ Ki-67 ↓ CD34 ↑ Bid |
[21] | |
| Athymic nude mice H1975 cells subcutaneously injected 5 × 106 cells/mouse |
UA—25 mg/kg−1 daily for 18 days |
↓ Tumor growth ↓ Tumor weight |
Not investigated | [14] | |
| Athymic Balb/c nude mice A549-PR cells |
UA 20 µM 72hr pre-injection co-culture |
↓ Tumorigenesis | Not investigated | [13] |
3. Ursolic Acid Derivatives and Their Effect against Lung Cancer
| Cell Line | Derivative Name | Derivative Structure | Findings | Mechanism | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 SF-295 (CNS) |
UA-9 10 nM |
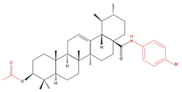 |
↓ Cell Density | Not investigated | [22] | |||||
| A549 | UA-triazolyl derivative | 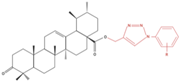 |
↓ Cell Density | Not investigated | [23] | |||||
| A549 | Compound 3B 50 µM, 48h | ↑ Apoptosis ↓ Cell viability ↓ Cell migration |
↓ NA+-K+-ATPase activity ↓ PKC activity ↓ VEGF protein ↓ ICAM-1 mRNA ↓ Fibronectin mRNA ↓ MMP 2 and 9 mRNA |
[5] | ||||||
 |
↓ Cell Growth | Not investigated | [ | 24 | ] | A549 | UA | |||
| H460 H322 | 5–20 µM 24 h |
↓ Proliferation ↓ Cell adhesion |
Compound 17 ↓ Wound healing ↓ Cell migration |
↑ E-cadherin ↓ N-cadherin ↓ vimentin ↓ AEG-1 ↓ NF-kB |
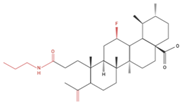 |
↑ Apoptosis ↑ Autophagy[6] |
||||
| ↑ Cleaved caspases 8 and 7 | ↑ Cleaved PARP | ↑ LC3A/B-II ratio ↓ Bcl-2 protein ↓ mTOR protein |
[25] | A549/H460 | UA | |||||
| A549 H460 | 30 µM 12, 24 and 48 h |
↓ Cell viability | UA232 24, 48 and 72 h ↓ Proliferation ↑ Apoptosis ↑ Chromatin condensation |
↑ Cleaved caspase 3/9 ↓ Bcl-2 ↑ Bax ↑ p-AMPK ↓ p-mTOR ↓ ACC activity |
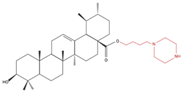 | ↓ FASN l activity |
↑ Cell cycle arrest [7] |
|||
| ↑ Apoptosis | ↓ Proliferation | ↓ Cyclin D1 protein | ↓ CDK4 protein ↑ CHOP protein ↑ Cleaved PARP |
[26] | A549 | UA 25, 50, 100, 250 and 500 µM |
↓ Cell viability | ↓ VRK1 autophosphorylation ↓ VRK1 activity ↓ p-CREB ↓ p-His-H3 | ||
| NCI-H460 | ↓ Cyclin D1 mRNA | 5Y8 | [8] | |||||||
| 5 and 10 µM | 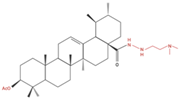 |
↑ Apoptosis G1 phase cell cycle arrest |
↓ p-NF-kB ↓ p-IKKα/β ↓ TAK1 ↓ TAB1 ↑ ROS |
[27] | PC9, H1299, A549, H1650, H358 and H1975 | UA 5, 10, 20, 30, 40, 50 and 80 µM 24, 48 and 72 h |
↓ Cell growth ↑ Apoptosis |
|||
| A549 | 8c 5, 10 and 20 µM 24 h | ↑ pSAPK/JNK | ↓ SP1 protein ↓ DNMT1 protein ↓ EZH2 protein |
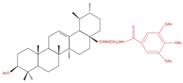 |
↑ Apoptosis G1 phase cell cycle arrest [9] |
|||||
| ↓ Cell migration | ↑ Caspase-3 cleavage | ↓ p-NF-kB | ↓ p-IKBα ↓ p-IKKα/β |
[28] | H28, H2452 and MSTO-211H | UA 0–80µM 24–72 h |
↑ Cytotoxicity ↓ Proliferation ↑ Sub-G1 population ↓ EMT |
↑ Cleaved caspase-3 ↑ Cleaved PARP ↑ E-cadherin ↓ N-cadherin ↓ β-catenin ↓ p-GSK2α/β ↓ cyclin D1 ↓ p-AKT ↓ NF-kB |
[10] | |
| A549 | UA 10–100 µM 24 h |
↓ Proliferation ↑ Apoptosis ↑ S-phase cell cycle arrest ↑ Autophagy |
↓ Bcl-2 protein ↑ Cleaved PARP ↑ LC3-II/LC3-I ratio ↑ p62 |
[11] | ||||||
| A549 | UA 11, 22, 44 and 88 µM 24 and 48 h |
↓ Cell viability ↑ Autophagy ↑ Mitophagy |
↑ LC3-II/LC3-I ratio ↑ p62 protein ↑ PINK1 protein ↓ p-AKT ↓ p-mTOR ↑ Nrf2 protein ↑ ROS |
[12] | ||||||
| A549 | UA 5, 10 and 20 µM 24, 48 and 72 h |
↓ Stemness ↓ Chemoresistance |
↓ CD133 ↓ Oct-4 ↓ Notch3 ↓ Nanog ↓ Sox2 |
[13] | ||||||
| H1975 NSCLC with EGFR T790M mutation |
UA 1, 5, 25, 50 and 100 µM |
↓ Cell growth ↑ Apoptosis ↓ Cell motility |
↓ CT45A2 mRNA ↓ TCF4 ↓ p-β-catenin @ Ser33/37/Thr41 ↑ p-GSK-3b @ Ser9 |
[14] | ||||||
| H1975 | UA 0.001–0.1 µM |
↓ EMT | ↓ N-cadherin ↑ E-cadherin ↓ MMP-2 and -9 ↓ TGF-β1 |
[15] | ||||||
| NCI-H292 | UA 3, 6, 9, 12 and 15 µM 24 and 48 h |
↓ Cell viability ↑ Apoptosis ↑ Ca2+ production ↓ Mitochondrial membrane potential |
↑ Cleaved caspase-7 ↑ Cleaved PARP ↑ Chromatin condensation ↑ Cytochrome c ↑ endo G ↑ AIF protein ↓ Bcl-2 protein ↓ BID protein |
[16] | ||||||
| A549, H460, H1975, H1299 and H520 H82 and H446 LLC |
UA 5–40 µM/ 48 h |
↓ Proliferation ↑ Apoptosis ↑ Autophagy ↓ Cell Viability |
↑ Cleaved PARP ↓ Bcl-2 protein ↑ LC3-II protein ↓ p-S6K @ T389 ↓ p-S6 @ S240-244 ↓ p-4E-BPI @ S65 ↓ p-AKT |
[17] | ||||||
| A549 and H460 | UA 10 and 20 µM |
↓ Proliferation ↑ Apoptosis ↑ G0/G1 cell cycle arrest ↓ Angiogenesis ↓ Migration ↓ Invasion ↓ Tumorsphere formation |
↓ p-EGFR ↓ VEGF ↓ MMP-2 ↓ PD-L1 ↓ CDK4 mRNA and protein ↓ CCND1 mRNA ↓ CCNE1 mRNA ↑ CDKN1A mRNA ↑ CDKN1B mRNA |
[18] | ||||||
| Human bronchial epithelial cells exposed to cigarette smoke extract | UA 3, 6, 12 and 25 µM |
↓ CSE-induced cytotoxicity | ↑ Nrf2 activity | [19] | ||||||
| H1299 | UA 50 and 80 µM 24 h |
↓ Cell survival ↑ Radiosensitivity |
↓ GSH (intracellular) ↓ HIF-1α protein |
[20] |
4. Patent Applications and Clinical Trials Related to Ursolic Acid Use
54. Conclusions
References
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. Proliferative Inhibition, Cell-Cycle Dysregulation, and Induction of Apoptosis by Ursolic Acid in Human Non-Small Cell Lung Cancer A549 Cells. Life Sci. 2004, 75, 2303–2316.
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting Mutant P53 for Efficient Cancer Therapy. Nat. Rev. Cancer 2018, 18, 89–102.
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-Suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015.
- Lai, M.Y.; Leung, H.W.C.; Yang, W.H.; Chen, W.H.; Lee, H.Z. Up-Regulation of Matrix Metalloproteinase Family Gene Involvement in Ursolic Acid-Induced Human Lung Non-Small Carcinoma Cell Apoptosis. Anticancer Res. 2007, 27, 145–153.
- Huang, C.-Y.; Lin, C.-Y.; Tsai, C.-W.; Yin, M.-C. Inhibition of Cell Proliferation, Invasion and Migration by Ursolic Acid in Human Lung Cancer Cell Lines. Toxicol. Vitr. 2011, 25, 1274–1280.
- Liu, K.; Guo, L.; Miao, L.; Bao, W.; Yang, J.; Li, X.; Xi, T.; Zhao, W. Ursolic Acid Inhibits Epithelial–Mesenchymal Transition by Suppressing the Expression of Astrocyte-Elevated Gene-1 in Human Nonsmall Cell Lung Cancer A549 Cells. Anti-Cancer Drugs 2013, 24, 494–503.
- Way, T.-D.; Tsai, S.-J.; Wang, C.-M.; Ho, C.-T.; Chou, C.-H. Chemical Constituents of Rhododendron Formosanum Show Pronounced Growth Inhibitory Effect on Non-Small-Cell Lung Carcinoma Cells. J. Agric. Food Chem. 2014, 62, 875–884.
- Kim, S.-H.; Ryu, H.G.; Lee, J.; Shin, J.; Harikishore, A.; Jung, H.-Y.; Kim, Y.S.; Lyu, H.-N.; Oh, E.; Baek, N.-I.; et al. Ursolic Acid Exerts Anti-Cancer Activity by Suppressing Vaccinia-Related Kinase 1-Mediated Damage Repair in Lung Cancer Cells. Sci. Rep. 2015, 5, 14570.
- Wu, J.; Zhao, S.; Tang, Q.; Zheng, F.; Chen, Y.; Yang, L.; Yang, X.; Li, L.; Wu, W.; Hann, S.S. Activation of SAPK/JNK Mediated the Inhibition and Reciprocal Interaction of DNA Methyltransferase 1 and EZH2 by Ursolic Acid in Human Lung Cancer Cells. J. Exp. Clin. Cancer Res. 2015, 34, 99.
- Sohn, E.J.; Won, G.; Lee, J.; Yoon, S.W.; Lee, I.; Kim, H.J.; Kim, S.-H. Blockage of Epithelial to Mesenchymal Transition and Upregulation of Let 7b Are Critically Involved in Ursolic Acid Induced Apoptosis in Malignant Mesothelioma Cell. Int. J. Biol. Sci. 2016, 12, 1279–1288.
- Lin, Y.-J.; Liang, W.-M.; Chen, C.-J.; Tsang, H.; Chiou, J.-S.; Liu, X.; Cheng, C.-F.; Lin, T.-H.; Liao, C.-C.; Huang, S.-M.; et al. Network Analysis and Mechanisms of Action of Chinese Herb-Related Natural Compounds in Lung Cancer Cells. Phytomedicine 2019, 58, 152893.
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444.
- Chen, Q.; Luo, J.; Wu, C.; Lu, H.; Cai, S.; Bao, C.; Liu, D.; Kong, J. The MiRNA-149-5p/MyD88 Axis Is Responsible for Ursolic Acid-mediated Attenuation of the Stemness and Chemoresistance of Non-small Cell Lung Cancer Cells. Environ. Toxicol. 2020, 35, 561–569.
- Yang, K.; Chen, Y.; Zhou, J.; Ma, L.; Shan, Y.; Cheng, X.; Wang, Y.; Zhang, Z.; Ji, X.; Chen, L.; et al. Ursolic Acid Promotes Apoptosis and Mediates Transcriptional Suppression of CT45A2 Gene Expression in Non-small-cell Lung Carcinoma Harbouring EGFR T790M Mutations. Br. J. Pharm. 2019, 176, 4609–4624.
- Ruan, J.S.; Zhou, H.; Yang, L.; Wang, L.; Jiang, Z.S.; Sun, H.; Wang, S.M. Ursolic Acid Attenuates TGF-Β1-Induced Epithelial-Mesenchymal Transition in NSCLC by Targeting Integrin AVβ5/MMPs Signaling. Oncol. Res. 2019, 27, 593–600.
- Chen, C.-J.; Shih, Y.-L.; Yeh, M.-Y.; Liao, N.-C.; Chung, H.-Y.; Liu, K.-L.; Lee, M.-H.; Chou, P.-Y.; Hou, H.-Y.; Chou, J.-S.; et al. Ursolic Acid Induces Apoptotic Cell Death Through AIF and Endo G Release Through a Mitochondria-Dependent Pathway in NCI-H292 Human Lung Cancer Cells In Vitro. Vivo 2019, 33, 383–391.
- Wang, M.; Yu, H.; Wu, R.; Chen, Z.; Hu, Q.; Zhang, Y.; Gao, S.; Zhou, G. Autophagy Inhibition Enhances the Inhibitory Effects of Ursolic Acid on Lung Cancer Cells. Int. J. Mol. Med. 2020, 46, 1816–1826.
- Kang, D.Y.; Sp, N.; Lee, J.-M.; Jang, K.-J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2021, 9, 297.
- Liu, W.; Tan, X.; Shu, L.; Sun, H.; Song, J.; Jin, P.; Yu, S.; Sun, M.; Jia, X. Ursolic Acid Inhibits Cigarette Smoke Extract-Induced Human Bronchial Epithelial Cell Injury and Prevents Development of Lung Cancer. Molecules 2012, 17, 9104–9115.
- Song, B.; Zhang, Q.; Yu, M.; Qi, X.; Wang, G.; Xiao, L.; Yi, Q.; Jin, W. Ursolic Acid Sensitizes Radioresistant NSCLC Cells Expressing HIF-1α through Reducing Endogenous GSH and Inhibiting HIF-1α. Oncol. Lett. 2017, 13, 754–762.
- Gao, Y.S.; Yuan, Y.; Song, G.; Lin, S.Q. Inhibitory Effect of Ursolic Acid and Oleanolic Acid from Eriobotrya Fragrans on A549 Cell Viability in Vivo. Genet. Mol. Res. 2016, 15, 1–8.
- Kalani, K.; Yadav, D.K.; Khan, F.; Srivastava, S.K.; Suri, N. Pharmacophore, QSAR, and ADME Based Semisynthesis and in Vitro Evaluation of Ursolic Acid Analogs for Anticancer Activity. J. Mol. Model 2012, 18, 3389–3413.
- Rashid, S.; Dar, B.A.; Majeed, R.; Hamid, A.; Bhat, B.A. Synthesis and Biological Evaluation of Ursolic Acid-Triazolyl Derivatives as Potential Anti-Cancer Agents. Eur. J. Med. Chem. 2013, 66, 238–245.
- Dar, B.A.; Lone, A.M.; Shah, W.A.; Qurishi, M.A. Synthesis and Screening of Ursolic Acid-Benzylidine Derivatives as Potential Anti-Cancer Agents. Eur. J. Med. Chem. 2016, 111, 26–32.
- Mendes, V.I.S.; Bartholomeusz, G.A.; Ayres, M.; Gandhi, V.; Salvador, J.A.R. Synthesis and Cytotoxic Activity of Novel A-Ring Cleaved Ursolic Acid Derivatives in Human Non-Small Cell Lung Cancer Cells. Eur. J. Med. Chem. 2016, 123, 317–331.
- Gou, W.; Luo, N.; Wei, H.; Wu, H.; Yu, X.; Duan, Y.; Bi, C.; Ning, H.; Hou, W.; Li, Y. Ursolic Acid Derivative UA232 Evokes Apoptosis of Lung Cancer Cells Induced by Endoplasmic Reticulum Stress. Pharm. Biol. 2020, 58, 707–715.
- Huang, R.-Z.; Hua, S.-X.; Liao, Z.-X.; Huang, X.-C.; Wang, H.-S. Side Chain-Functionalized Aniline-Derived Ursolic Acid Derivatives as Multidrug Resistance Reversers That Block the Nuclear Factor-Kappa B (NF-ΚB) Pathway and Cell Proliferation. Med. Chem. Commun. 2017, 8, 1421–1434.
- Jiang, W.; Huang, R.-Z.; Zhang, J.; Guo, T.; Zhang, M.-T.; Huang, X.-C.; Zhang, B.; Liao, Z.-X.; Sun, J.; Wang, H.-S. Discovery of Antitumor Ursolic Acid Long-Chain Diamine Derivatives as Potent Inhibitors of NF-ΚB. Bioorg. Chem. 2018, 79, 265–276.
- Yin, M.-C.; Lin, M.-C.; Mong, M.-C.; Lin, C.-Y. Bioavailability, Distribution, and Antioxidative Effects of Selected Triterpenes in Mice. J. Agric. Food Chem. 2012, 60, 7697–7701.
- Yang, L.; Sun, Z.; Zu, Y.; Zhao, C.; Sun, X.; Zhang, Z.; Zhang, L. Physicochemical Properties and Oral Bioavailability of Ursolic Acid Nanoparticles Using Supercritical Anti-Solvent (SAS) Process. Food Chem. 2012, 132, 319–325.
- Wang, W.; Zhang, W.; Jiang, Y.; Wang, X.; Zhang, X.; Liu, H.; Zhang, T. Preparation of Ursolic Acid–Phospholipid Complex by Solvent-Assisted Grinding Method to Improve Dissolution and Oral Bioavailability. Pharm. Dev. Technol. 2020, 25, 68–75.
- Xia, Y.; Wei, G.; Si, D.; Liu, C. Quantitation of Ursolic Acid in Human Plasma by Ultra Performance Liquid Chromatography Tandem Mass Spectrometry and Its Pharmacokinetic Study. J. Chromatogr. B 2011, 879, 219–224.
- Ramírez-Rodríguez, A.M.; González-Ortiz, M.; Martínez-Abundis, E.; Acuña Ortega, N. Effect of Ursolic Acid on Metabolic Syndrome, Insulin Sensitivity, and Inflammation. J. Med. Food 2017, 20, 882–886.
- Wang, X.-H.; Zhou, S.-Y.; Qian, Z.-Z.; Zhang, H.-L.; Qiu, L.-H.; Song, Z.; Zhao, J.; Wang, P.; Hao, X.-S.; Wang, H.-Q. Evaluation of Toxicity and Single-Dose Pharmacokinetics of Intravenous Ursolic Acid Liposomes in Healthy Adult Volunteers and Patients with Advanced Solid Tumors. Expert Opin. Drug Metab. Toxicol. 2013, 9, 117–125.