Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Eric O'Neill and Version 2 by Beatrix Zheng.
The pancreas is a glandular organ with endocrine and exocrine functions necessary for the maintenance of blood glucose homeostasis and secretion of digestive enzymes. Pancreatitis is characterized by inflammation of the pancreas leading to temporary or permanent pancreatic dysfunction. Inflammation and fibrosis caused by chronic pancreatitis exacerbate malignant transformation and significantly increase the risk of developing pancreatic cancer, the world’s most aggressive cancer with a 5-year survival rate less than 10%. Berberine (BBR) is a naturally occurring plant-derived polyphenol present in a variety of herbal remedies used in traditional medicine to treat ulcers, infections, jaundice, and inflammation.
- berberine
- polyphenols
- pancreatic cancer
- apoptosis
1. Effects of Berberine against Pancreatic Cancer: In Vitro Studies
Treatment of BxPC-3 human pancreatic adenocarcinoma cells and HPDE-E6E7c7 normal human pancreatic ductal epithelial cells with BBR (10–200 µM) for 24–72 h showed that high concentration of BBR can inhibit pancreatic cancer cell growth and trigger caspase-independent cell death [1][108]. BBR (10–200 µM) treatment for 24–72 h caused a concentration and time-dependent reduction in cell proliferation and BxPC-3 cells were more sensitive to the cytotoxic effect of BBR after 24 h than HPDE-E6E7c7 cells. BBR (150–200 µM) treatment significantly increased caspase-3- and-7 activity in both cell lines (although activation was much greater in HPDE-E6E7c7 cells) and caspase inhibition using Z-VAD-FMK rendered cells less susceptible to BBR, suggesting that BBR induces apoptosis of cancer cells at high concentrations [1][108]. Immunostaining showed that treatment with 200 µM BBR for 24 h caused translocation of apoptosis-inducing factor (AIF), a caspase-independent death effector, from the mitochondria to the nucleus indicating that BBR also induces caspase-independent mechanisms of apoptosis. Visualisation of live cells treated with fluorescent BBR for 24 h showed that at low concentrations (10–50 µM) BBR is localised to the mitochondria, but at higher concentrations (100–200 µM) BBR localisation extends into the cytoplasmic and nuclear compartments [1][108] (Table 12).
Lovastatin is a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor that blocks cholesterol synthesis and is predominately used to treat cardiovascular disease. Panc02 cells were treated with increasing concentrations of BBR (0–5 µM) or lovastatin (0–0.12 µM) for 48 h, alone and in combination, and cell viability was assessed with crystal violet staining [2][109]. Both drugs showed dose-dependent inhibition of cell viability and the combination of both drugs had highly synergistic cytostatic/cytotoxic effects. BBR or lovastatin treatment alone did not affect cell cycle distribution, but combined treatment resulted in a two-fold increase in the percentage of cells in sub-G1 phase and a modest increase in the percentage of cells in G1 phase. Interestingly, pre-treatment with products of the mevalonic acid pathway (i.e., restoring cholesterol synthesis) attenuated the anticancer effects of lovastatin but not BBR, indicating that BBR enhances the anticancer effects of lovastatin independent of the cholesterol synthesis pathway [2][109] (Table 12). Additionally, inhibiting farnesyltransferase or geranylgeranyltransferase did not potentiate the effects of BBR further, suggesting that the effects of BBR against pancreatic cancer do not rely on inhibition of protein prenylation. Overall, this researchtudy indicates that BBR can slow the growth of pancreatic cancer cells in vitro through a mechanism that does not involve the cholesterol synthesis pathway.
BBR treatment of PDAC cells (PANC-1 and MiaPaCa-2) for 17–72 h inhibited proliferation and DNA synthesis, and increased the population of cells in the G1-phase of the cell cycle with a concomitant reduction in the S and G2/M-phase population [3][110]. Furthermore, BBR caused a decrease in mitochondrial membrane potential and a concentration-dependent decrease in ATP levels to a similar degree as metformin, an established mitochondrial complex I inhibitor with tumour-suppressive effects [3][4][110,111].These changes in mitochondrial function coincided with a concentration-dependent increase in phosphorylated levels of ACC (Ser79) and AMPK (Thr172), suggesting that BBR supresses the growth of PDAC cells through a mechanism involving decreased mitochondrial function leading to decreased ATP levels and activation of AMPK. BBR blocked neurotensin- and insulin-induced ERK and mTORC1 activation in a concentration-dependent manner as indicated by decreased phosphorylation of ERK (Thr202/Tyr204), p70S6k (Thr389), and S6 (Ser240/244). Interestingly, siRNA knockdown of AMPK blocked the effects of low dose BBR (<3µM) and only partially blocked the effects of higher concentrations (3–6 µM). Overall, these data provide evidence that BBR impairs mitochondrial function of PDAC cells and the effects of BBR against PDAC rely on AMPK-dependent and independent mechanisms (Table 12).
Cancer stem cells (CSCs) play a major role in the initiation, growth, and metastasis of tumours since they have similar characteristics to ordinary stem cells including multipotentiality and high capacity for self-renewal [5][112]. One method for identifying CSCs is to identify side population (SP) cells based on their ability to exclude Hoeschst dye, a characteristic associated with stemness [5][112]. Treatment of PANC-1 cells with BBR decreased the proportion of SP cells from 9.7 to 5.7% and this corresponded with down-regulation of stem cell-associated genes: SOX2, OCT4, and NANOG [6][113]. MIA-PaCa-2 cells had no SP cells in the control group but still had down-regulation of stem cell-associated genes following treatment with BBR [6][113] (Table 12). Overall, these results indicate that BBR may reduce initiation, growth, and metastasis of tumours by reducing stemness of pancreatic cancer cells.
Treatment of PANC-1 and Mia-PaCa-2 pancreatic cancer cells with 1–15 µM BBR for 72 h resulted in concentration-dependent inhibition of cell growth with IC50 values of 15 µM and 10 µM, respectively [7][114]. BBR significantly increased the G1 phase population of PANC-1 cells with a concomitant reduction in S phase population. Additionally, BBR treatment induced apoptosis of PANC-1 and Mia-PaCa-2 cells as indicated by increased Annexin V/PI staining and increased caspase-3/7 activity with 24 h and 48 h treatment but not 72 h treatment [7][114]. The proapoptotic effect of BBR coincided with a concentration-dependent increase in intracellular ROS levels suggesting that the effects of BBR may be ROS-dependent [7][114] (Table 12).
BBR was found to localize to the cytoplasm of MiaPaCa-2 cells following 1 h treatment with 10 µM concentration, and at 50 µM or 150 µM concentrations BBR is also found in the nucleus [8][115]. Furthermore, the localization of BBR is maintained after 48 h of treatment, and 48 h treatment with BBR (0.4–50 µM) caused a concentration-dependent reduction in cell viability. Use of the mitochondrial tracer tetramethyl rhodamine methyl ester (TMRM) showed that BBR localizes to the mitochondria and 50 µM BBR (48 h) was found to decrease citrate synthase activity, suggesting that BBR impairs mitochondrial function [8][115]. Additionally, 10 µM BBR (48 h) was sufficient to induce G1 cell cycle arrest and significantly decrease the S phase population. Furthermore, 48 h treatment with 10 µM and 50 µM caused a 20- and 33-fold increase in mRNA expression of cyclin dependent kinase inhibitor 1A (P21)—a marker of cellular senescence—and this coincided with a concomitant increase in senescence-associated β-galactosidase staining and increase in apoptotic caspase-3 activity [8][115]. BBR treatment was also found to induce autophagy of Mia-PaCa-2 cells as indicated by increased mRNA expression of LC3 and Beclin-1, and increased protein expression of LC3-I and LC3-II [8][115]. BBR treatment of Mia-PaCa-2 cells also decreased cell migration in a wound-healing assay and decreased invasion in a transwell assay which coincided with decreased mRNA expression of C-X-C motif chemokine receptor 4 (CXCR4) involved in migration and increased mRNA expression of death-associated protein 1 (DAP1) [8][115]. DNA methyltransferases (DNMT) epigenetically regulate enzymes involved in DNA repair mechanisms [9][116]. BBR treatment upregulated mRNA expression of DNMT1, DNMT3A, DNMT3B, and O6-methylguanine DNMT (MGMT) [8][115] (Table 12).
Treatment of Panc-1 pancreatic cancer cells with BBR (1–30 µM) for 72 h caused a concentration-dependent decrease in cell viability with an IC50 of 4.76 µM and treatment with 10 µM BBR for 48 h caused significant inhibition of cell metastasis in a transwell assay [10][117]. BBR treatment significantly decreased expression of tumour necrosis factor α (TNFα), carbohydrate antigen 242 (CA242, a diagnostic and poor prognostic biomarker of pancreatic cancer [11][118]), and the oncogenic protein K-Ras. Conversely, BBR upregulated the expression of the tumour suppressor cyclin dependent kinase inhibitor 2A (CDKN2A). Metabolomic analysis revealed that BBR dysregulates pancreatic cancer cell metabolism with a similar regulatory pattern to the pancreatic cancer drug gemcitabine, but the effect of BBR on cell metabolism was much stronger than that of gemcitabine. Additionally, BBR increased energetic metabolism (i.e., glycolysis and glutamine)-associated metabolites and decreased citric acid cycle-associated metabolites. Transmission electron microscopy confirmed that these metabolic changes were due to BBR-induced mitochondrial damage and this damage coincided with decreased citrate metabolism [10][117] (Table 12). Overall, these data indicate that BBR inhibits pancreatic cancer cell viability through a mechanism that likely involves mitochondrial damage leading to decreased citrate metabolism and disruption of fatty acid biosynthesis, which has an important role in the proliferation and metastasis of pancreatic cancer cells.
BBR treatment (1–2000 nM, 96 h–2 weeks) inhibited colony formation of MIA-PaCa-2 pancreatic cancer cells and inhibited the viability of MIA-PaCa-2, PANC-28, and AsPC-1 in a concentration-dependent manner with IC50 values of 1700 nM, 2000 nM, and 2000 nM, respectively [12][119]. MIA-PaCa-2 cells possess a R248W TP53 gain-of-function mutation resulting in expression of a p53 protein with a mutated DNA binding domain effectively blocking the tumour suppressive properties of p53 [13][120]; transfecting MIA-PaCa-2 cells to express wildtype p53 caused a three-fold reduction in the IC50 of BBR [14][121]. Furthermore, BBR sensitized MIA-PaCa-2 cells to the MDM2 inhibitor Nutlin-3a [15][122]. MIA-PaCa-2 cells transfected with kinase-dead GSK-3β had more than three-fold greater sensitivity to BBR-induced inhibition of cell viability compared to cells expressing wildtype GSK-3β [16][123]. Similarly, BBR decreased colony formation to a much greater extent in kinase-dead GSK-3β mutants compared to cells expressing wildtype GSK-3β [16][123]. Overall, these studies provide evidence of the effectiveness of BBR against pancreatic cancer in vitro and suggest that expression of wildtype p53 and/or inhibition of MDM2 renders pancreatic cancer cells more sensitive to BBR, and that GSK-3β may be an important regulator of the sensitivity of pancreatic cancer cells to BBR (Table 12).
In another study, BBR (0–30 µM; 24 h) had no effect on proliferation and migration of pancreatic cancer cells (AsPC-1 and SW1990); however, BBR (3–30 µM; 20 h) did inhibit trans-endothelial migration of AsPC-1 cells, indicating that BBR may exert its anticancer effects by improving the lung vascular barrier [17][124]. TGF-β1 is involved in disruption of the endothelial barrier and its receptor (TGFBR1) is a predicted target of BBR based on motif-based screening in pharmacophore databases [18][19][125,126]. Pancreatic cancer cells (PANC-1, SW1990, and AsPC-1) expressed higher levels of TGF-β1 compared to normal pancreatic ductal epithelial cells, and inhibition of TGFBR1 in endothelial cells blocked the inhibitory effect of BBR on trans-endothelial migration of AsPC-1 cells [17][124]. Additionally, BBR caused a concentration-dependent decrease in levels of phosphorylated smad3, phosphorylated smad2, SNAIL1, and SLUG in endothelial cells, which are all downstream targets of TGF-β1 signalling. Using surface plasmon resonance and molecular docking studies, it was determined that BBR binds to TGFBR1 with an equilibrium dissociation constant of 18.0 µM, BBR inhibits TGFBR1 kinase activity in a concentration-dependent manner with an IC50 of 7.056 µM, and that BBR docks into the active site of TGFBR1 interacting with key residues including Glu45, Tyr49, Asp81, Tyr82, and His83 [17][124]. Overall, these data indicate that BBR reduces metastasis of pancreatic cancer cells by interacting with the intracellular kinase domain of TGFBR1 to prevent TGF-β1-induced damage to endothelial barrier (Table 12).
Primary acinar cells were treated with TGF-β (5 ng/mL; 2 days) to induce acinar-to-ductal metaplasia (ADM) and concurrently (2 days) or subsequently (1 day) treated with 10 µM BBR [20][127]. BBR attenuated induction of ADM in TGF-β-treated cells, increased levels of amylase, and decreased CK19 levels closer to basal [20][127]. In the early stages of PanIN, but before development of PDAC, there is a metabolic shift towards glycolysis known as the Warburg effect [21][128]. TGF-β-treated acinar cells had increased glucose consumption and lactate production, markers of glycolysis, but BBR restored these levels to basal and the glycolysis-supressing effect of BBR was comparable to 2-deoxy-D-glucose (2-DG), a glycolysis inhibitor [20][127]. Additionally, both BBR and 2-DG decreased glycolysis-related gene expression: lactate dehydrogenase (LDHA), aldolase A (ALDOA), live phosphofructokinase (PFKL), pyruvate kinase M2 (PKM2), and 3′-phosphoinositide-dependent kinase 1 (PDK1). Interestingly, BBR increased activated levels of AMPK, and decreased levels of active mTOR and HIF-1α [20][127]. Compound c, an AMPK inhibitor, restored glycolysis and prevented BBR from suppressing the development of PanIN [20][127]. Taken together, these results indicate that BBR can prevent and, to some extent, reverse PanIN due to CP by activating AMPK and supressing the Warburg effect. Furthermore, treatment of MIN6 insulinoma cells with BBR (2.5–50 µM, 2–24 h) resulted in concentration-dependent reduction in cell viability with an IC50 of 5.7 µM for 16 h treatment and this coincided with a concentration-dependent increase in the population of apoptotic cells as indicated by increased Annexin V staining and increased DNA fragmentation [22][129]. These effects of BBR coincided with increased levels of pro-apoptotic proteins including increased cytoplasmic cytochrome C, AIF, Apaf-1, Bax, cleaved caspase-3, and cleaved PARP, and decreased expression of the antiapoptotic marker Bcl-2 [22][129] (Table 12). Ultimately, these data show that BBR stimulates apoptosis of insulinoma cells.
Table 12.
Effects of BBR against pancreatic cancer in vitro.
| Model | Treatment | Effects | Signalling | Ref. | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BxPC-3 | 10–200 µM BBR 24–72 h |
↓ Proliferation | Synergistic effect | ↑ Caspase 3/7 | [2][1] | [108] | |||||||||||||
| [ | 109 | Panc02 | 0–10 µM BBR 0–0.12 µM Lovastatin 48 h |
↓ Cell viability | - | [2] | [109] | ||||||||||||
| ↑ Sub-G | 1 | and G | 1 | phase populations | |||||||||||||||
| PANC-1 MiaPaCa-2 |
0.3–6 µM BBR 17–72 h |
↓ DNA synthesis | ↑ pAMPK (Thr | 172 | ) ↑ pACC (Ser | 79 | ) ↓ mTORC1 ↓pp70 S6K (Thr | 389 | ) ↓ pS6 (Ser | 240/244 | ) ↓ pERK (Thr | 202 | /Tyr | 204 | ) ↑ pRaptor (Ser | 792 | ) | [3] | [110] |
| ↓ Proliferation | |||||||||||||||||||
| ↑ G | 1 | -phase population | |||||||||||||||||
| ↓ S and G | 2 | /M-phase population | |||||||||||||||||
| ↓ Mitochondrial membrane potential |
|||||||||||||||||||
| ↓ ATP levels | |||||||||||||||||||
| Mia-PaCa-2 PANC-1 |
15 µM BBR 72 h |
↓ CSC population | ↓ SOX2 ↓ OCT4 ↓ NANOG |
[6] | [113] | ||||||||||||||
| ] | |||||||||||||||||||
| Intraperitoneal injection 30 mg/kg/day |
|||||||||||||||||||
| BBR: Oral administration 100 mg/kg/day for 14 days |
|||||||||||||||||||
| MiaPaCa-2 xenografted nude mice | Intraperitoneal injection | ↓ Tumour weight ↓ Tumour volume |
[3] | [110] | |||||||||||||||
| 5 mg/kg/day BBR | |||||||||||||||||||
| AsPC-1 intravenous injection (tail vein) in BALB/c nude mice | Oral Gavage | PANC-1 Mia-PaCa-2 |
1–15 µM BBR 72 h |
↓ Cell viability | ↑ Caspase-3/7 activity | [7] | [114] | ||||||||||||
| ↑ G | 1 | -phase population | |||||||||||||||||
| ↓ S-phase population | |||||||||||||||||||
| ↑ Survival | ↓ Lung metastases ↓ Lung infiltration |
[17] | [124] | ||||||||||||||||
| 100–200 mg/kg/day BBR | |||||||||||||||||||
| Beginning 3 days prior to AsPC-1 injection | |||||||||||||||||||
| Cerulein-induced CP/CP-induced neoplasia C57BL/6 mice |
Intragastric Administration | ↓ ADM ↓ PanIN ↓ Fibrosis ↓ CK19 ↑ Amylase |
[20] | [127] | ↑ Apoptosis | ||||||||||||||
| ↑ ROS | |||||||||||||||||||
| Mia-PaCa-2 | |||||||||||||||||||
| BBR 200 mg/kg | 10–50 µM BBR 1–48 h |
↓ Cell viability | ↑ p21 ↑ Caspase-3 activity ↑ LC3 ↑ DAP1 ↓ CXCR4 ↑DNMT1 ↑DNMT3A ↑ DNMT3B ↑ MGMT |
[8 | |||||||||||||||
| Daily, 3 days/week | ] | [ | 115 | ] | |||||||||||||||
| BBR mitochondrial localisation | |||||||||||||||||||
| ↓ Citrate synthase activity | |||||||||||||||||||
| ↑ G | 1 | -phase population | |||||||||||||||||
| ↓ S and G | |||||||||||||||||||
| 4–8 weeks | 2 | -phase population | |||||||||||||||||
| ↑ Senescence | |||||||||||||||||||
| ↓ Migration | |||||||||||||||||||
| ↓ Invasion | |||||||||||||||||||
| Panc-1 | 1–60 µM BBR 48–72 h |
↓ Cell viability | ↓ TNFα ↓ CA242 ↓ K-Ras ↑ CDKN2A |
[10] | [117] | ||||||||||||||
| ↑ Apoptosis | |||||||||||||||||||
| ↓ Metastasis | |||||||||||||||||||
| ↑ Glycolysis-associated metabolites |
|||||||||||||||||||
| ↑ Glutamine-associated metabolites |
|||||||||||||||||||
| ↓ Citric acid cycle-associated metabolites |
|||||||||||||||||||
| ↑ Mitochondrial damage | |||||||||||||||||||
| ↓ Citrate metabolism | |||||||||||||||||||
| MIA-PaCa-2 PANC-28 AsPC-1 |
1–2000 nM BBR 72 h–2 weeks |
↓ Cell viability | - | [12][14][15][16] | [119,121,122,123] | ||||||||||||||
| ↓ Colony formation | |||||||||||||||||||
| PANC-1 AsPC-1 SW1990 |
0–30 µM BBR 24 h |
↓ Trans-endothelial migration | ↓ pSmad2 ↓ pSmad3 ↓ SNAIL1 ↓ SLUG |
[17] | [124] | ||||||||||||||
| TGF-β-treated Primary acinar cells | ADM induction: 5 ng/mL TGF-β 2 Days 10 µM BBR 1–2 days |
↓ PanIN | ↓ CK19 ↓ LDHA ↓ALDOA ↓ PFKL ↓ PKM2 ↓ PDK1 ↑ pAMPK ↓ pmTOR ↓ HIF-1α |
[20] | [127] | ||||||||||||||
| ↓ ADM | |||||||||||||||||||
| ↓ Glycolysis | |||||||||||||||||||
| MIN6 | 2.5–50 µM BBR 2–24 h |
↓ Cell viability | ↑ Cytochrome C ↑ AIF ↑ Apaf-1 ↑ Bax ↑ Cleaved Caspsase-3 ↑ Cleaved PARP ↓ Bcl-2 |
[22] | [129] | ||||||||||||||
| ↑ Apoptosis | |||||||||||||||||||
| ↑ DNA fragmentation |
Table legend: ↑ increases, ↓ reduces.
2. Effects of Berberine against Pancreatic Cancer: In Vivo Studies
Treatment of Panc02 xenografted C57/B16 mice with BBR (oral administration, 100mg/kg/day) or lovastatin (intraperitoneal injection, 30 mg/kg/day) for 14 days slightly decreased tumour volume when given alone, but significantly decreased tumour volume when given in combination [2][109] (Table 23). Ultimately, this researchtudy shows that BBR administered orally reaches high enough concentrations in vivo to slow tumour growth and that BBR acts synergistically with cholesterol-lowering medications such as lovastatin.
Table 23.
Effects of BBR against pancreatic cancer in vivo.
| Animal Model | Treatment | Effects | Ref. |
|---|---|---|---|
| Panc02 xenografted c57Bl/6 mice | Lovastatin: | ↓ Tumour volume |
Table legend: ↑ increases, ↓ reduces.
Mice that underwent intragastric administration of BBR (100–200 mg/kg/day) had a dose-dependent increase in overall survival after being intravenously injected with AsPC-1 cells through the tail vein [17][124]. Furthermore, the mice administered BBR (200 mg/kg/day) had 36.5% fewer lung metastases and 35% lower overall area of metastatic nodes in the lungs after six weeks. Ki-67 staining of lung metastases showed no changes between BBR-treated and control mice, indicating that BBR did not significantly affect the proliferation of tumour cells in lung metastases. In order to assess lung infiltration of circulating tumour cells, researchers labelled AsPC-1 cells with carboxyfluorescein succinimidyl ester (CFSE) prior to injecting them into the tail vein of mice; 24 h later, the lungs were homogenized to create a single cell suspension that was analysed using flow cytometry which revealed that BBR reduced accumulation of pancreatic cancer cells in the lungs, and 3D reconstruction fluorescent images of lung tissue sections showed that the cancer cells that did accumulate in the lungs were outside of the microvessels, suggesting that BBR may block transportation of tumour cells from circulation into the interstitial fluid [17][124]. As mentioned in the previous section, TGFBR1 was found to play a crucial role in the antimetastatic effect of BBR in vitro, and inhibition of TGFBR1 activation in vivo using A83-01 (10 mg/kg/day) also suppressed the antimetastatic effect of BBR [17][124] (Table 23).
Induction of CP in C57BL/6 mice using cerulein (50 µM/kg, 6 h/day, 3 days/week, 8 weeks) resulted in acinar-to-ductal metaplasia (ADM) after 4 weeks and pancreatic PanIN after 8 weeks [20][127]. BBR treatment (200 mg/kg/day, 3 days/week) for 4 weeks following induction of CP decreased the area of preneoplastic lesions. BBR given for 8 weeks alongside cerulein had a greater therapeutic effect than BBR administered after establishment of CP as indicated by inhibition of the development of ADM, PanIN, and fibrosis [20][127]. Mice with CP had decreased levels of serum amylase which is characteristic of damaged acinar cells in PanIN lesions [23][130], however, BBR treated mice had amylase levels closer to basal [20][127] (Table 23). Overall, these results show that BBR can prevent and partially reverse neoplastic lesions in the pancreas caused by CP.
Several studies have investigated the effects of BBR in PanIN. In addition to inhibiting proliferation and survival of pancreatic cancer cells, BBR treatment was able to mitigate many of the cellular changes that are observed in PanIN. Notably, BBR restored p16 and p21 function, and inhibited oncogenic KRAS as well as downstream effectors of KRAS signalling including ERK, mTOR, and p70S6K (Figure 15). Additionally, BBR treatment activated AMPK signalling and initiated mitochondrial-mediated apoptosis. In animal models, BBR administered intraperitoneally or intragastrically inhibited tumour growth, decreased tumour metastasis, and prevented pancreatitis-induced malignant transformation. Taken together, these findings suggest significant effects of BBR against pancreatic cancer and point to the need of more animal studies and future clinical trials.
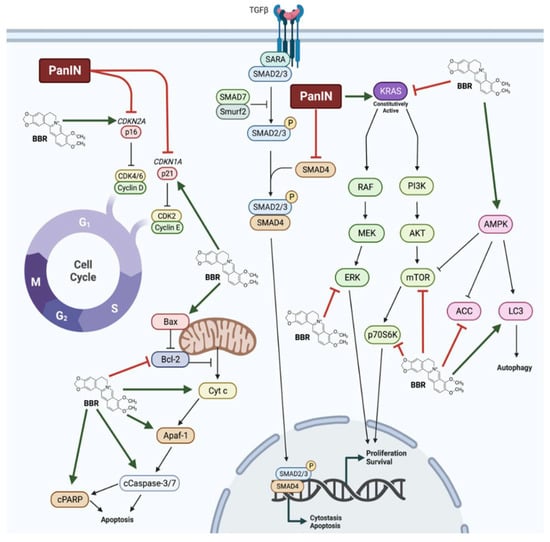
Figure 15. Summary of the effects of berberine in pancreatic cancer cells. Berberine (BBR) was shown to restore p16 and p21 function in pancreatic intraepithelial neoplasia (PanIN). BBR increased AMP-activated protein kinase (AMPK) and apoptosis signalling and decreased oncogenic Kirsten rat sarcoma virus (KRAS), extracellular signal-regulated kinase (ERK), mammalian target of rapamycin (mTOR), and ribosomal protein S6 kinase (p70S6K) signalling, culminating in decreased proliferation and survival, and increased apoptosis, cytostasis, and autophagy.