Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Daniele Bottai.
Neural stem cells (NSCs) in the adult nervous tissue were among the last to be identified and isolated in a living organism. While the U.S. Food and Drug Administration and the European Medicines Evaluation Agency have recently approved new drugs to treat spinal muscular atrophy 1 (SMA1) in young patients, they are mostly ineffective in older patients since many motor neurons have already been lost. Therefore, understanding nervous system (NS) physiology in SMA patients is essential.
- spinal muscular atrophy
- induced pluripotent stem cells
- differentiation
1. Epigenetic
Epigenetic DNA mechanisms include the frequent methylation of cytosine residues’ five positions, inhibiting neighboring gene expression. Common histone modifications include acetylation, ubiquitination, and methylation of lysine or arginine residues in the histone tails, either promoting or repressing gene expression.
While DNA methylation patterns are relatively stable in terminally differentiated cells, they are strikingly diverse among different tissue stem cell types and change dynamically during development. Whether methylation levels among SMN2 copies influence NSC proliferation or differentiation remains unclear. Among the various DNA (cytosine-5)-methyltransferase (Dnmt) families [89][1]), the Dnmt3 family contains at least two essential members with nonoverlapping functions in development: Dnmt3a and Dnmt3b. Their inactivation eliminated de novo methylation in mouse embryos, causing death four weeks after birth (Dnmt3a) or embryonic lethality (Dnmt3b) [90][2], and increasing NSC proliferation.
DNMT3a and DNMT3b catalyze the de novo addition of methyl groups to DNA, while DNMT1 maintains methylation patterns in newly synthesized DNA [91][3]. Dnmt3 orthologs have a variable N-terminal region (~280 aa in Dnmt3a and ~220 aa in Dnmt3b) in many species, including humans and mice. Nevertheless, they all contain a PWWP motif [92][4]. Detailed PWWP sequence analysis indicated that this domain comprises a five-stranded β-barrel structure similar to the SMN Tudor domain [93][5]. However, the structural similarity between Tudor and PWWP domains remains uncertain.
Non-CG DNA methylation (mCH; where H = A, C, or T) is enriched in mouse and human neurons compared with other cell types. It occurs primarily at cytosines preceding an adenine (mCA). The mCH rate is lower than the mCG rate. Conversely, in some neuron classes, the number of modified CH sites surpasses those of modified CG sites [94][6].
Dnmt3a catalyzes the de novo modification of mCG and mCH sites. Its expression is upregulated in neurons starting at birth and peaking at 2 weeks before plateauing by 4–6 weeks in the frontal cortex and declining in adulthood in the mouse [95,96][7][8]. Full mCH accumulation takes 16 years in humans, even though most sites are formed in the first 2 years. Furthermore, it has a heterogeneous distribution with no mCH deposition at completely silent genes or inaccessible constitutive heterochromatin regions but appreciable accumulation at extragenic regions, repeated sequences, inactive regulatory elements, and lowly transcribed genes. However, DNMT3A binding and mCH accumulation are missing from highly expressed genes and active regulatory elements [97][9].
At a local (kilobase) scale, mCH exhaustion at genes and regulatory elements mostly aligns with mCG patterns. Nevertheless, mCH shows distinctive large (megabase) scale patterns most likely associated with chromosome folding within the nucleus and topologically associating domains in chromatin folding [98][10].
The association between gene transcription and chromatin folding is undoubtedly associated with mCH deposition. While the mechanisms regulating Dnmt3a remain unclear, the analysis of different histone modifications in mouse cortex indicates that mCH deposition is controlled by chromatin structure, especially during early postnatal development [99][11].
Non-CG methylation is known to be highly cell-type specific, either at local or global levels. For example, in mice and humans, mCH levels can vary by up to 2-fold between brain regions [94,95,96][6][7][8], 1.5-fold among neuron subtypes present in the same brain region [100[12][13],101], and increases in cortical excitatory neurons from the upper to deeper layers [102][14].
DNMT3a expression decreased through various differentiation phases (i.e., dorsal fate specification, neural progenitor cells [NPCs], self-organized rosettes, and maturing neuronal cells) in an iPSC corticogenesis model [103][15]. Presumably, the methylation level of various genes also decreases within those differentiation phases.
SMN2 is also subject to silencing by DNA methylation. Indeed, SMN2 contains four CpG islands with conserved methylation patterns. They contain 85 CpG dinucleotides, 14 of which are between nts −896 and −645, 12 between nts −469 and −247, 38 between nts −151 and 296, and 21 between nts 844 and 1146.
Fibroblast cell lines from SMA patients showed almost complete methylation at CpG1 and CpG4, while CpG2 was partially methylated and CpG3 showed very little methylation [104][16]. CpG methylation (at positions −290 and −296) in blood-borne cells correlated with disease severity and first transcriptional start site activity at SMN2 at position −296 in patients affected by severe SMA and suffering from mild SMA but carrying identical SMN2 copy numbers [104][16]. Inhibiting SMN2 silencing via DNA methylation is considered a promising pharmacologic SMA therapy [104,105][16][17].
SMN2 expression does not change significantly during healthy cell development and differentiation. This observation is expected since SMN1 is the primary SMN gene expressed in healthy individuals. However, SMN1 expression showed the same pattern [103][15].
Nevertheless, a deeper analysis showed some differences in SMA neural development. SMN2 showed hypomethylation in early development (iPSCs, neuroepithelial precursors (NEP), and immature (IMN) and mature (MMN) MNs) that most likely reflects its increased expression in cells derived from SMA1 patients than from SMA2 patients or healthy individuals to compensate for SMN1′s absence (Figure 1) [106][18].
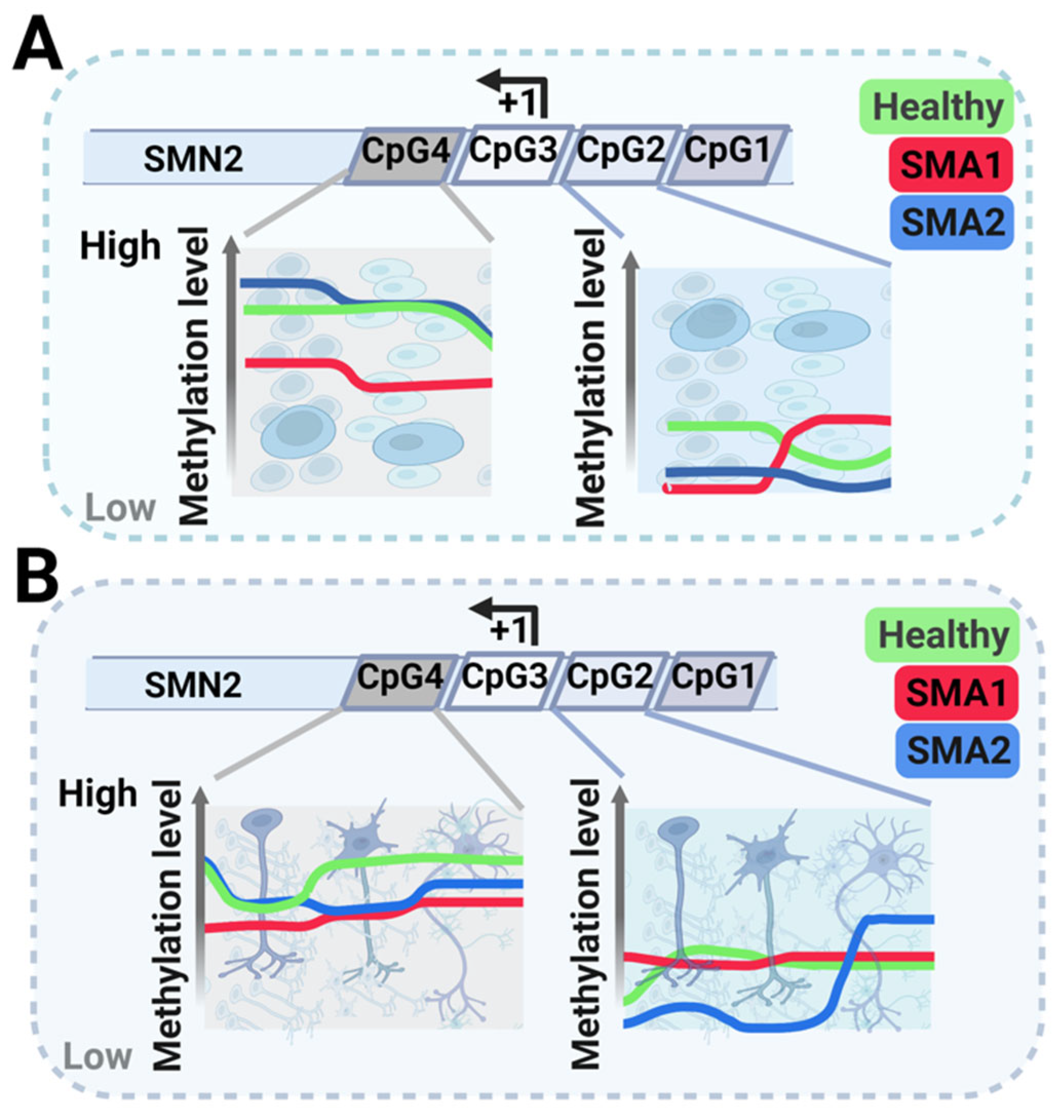
Figure 1. Schematic representation of methylation levels in the survival of motor neuron 2 (SMN2) gene at various differentiation stages during motor neuron (MN) development: induced pluripotent stem cells (iPSCs), neuroepithelial precursors (NEP) in panel (A), immature MNs (IMN), and mature MNs (MMNs) in panel (B). Green represents healthy individuals, blue represents SMA2 patients, and red represents SMA1 patients. The scheme also describes methylation in the SMN2 gene’s four different CpG islands.
More specifically, the methylation at CpG2 varied from stage to stage with no clear tendency (Figure 1). However, while CpG4 methylation in SMA1 patient-derived cells was significantly lower at most development stages, it was significantly lower in the final two MN differentiation stages (IMM and MMN) in SMA2 patient-derived cells (Figure 1B).
The same study [106][18] compared the DNA methylation of genes involved in differentiation from iPSCs to NEP to MNs, starting from genes involved in maintaining the pluripotency state particularly relevant to iPSCs. OCT4 is commonly hypomethylated in ESCs or iPSCs [107,108][19][20]. However, it showed greater methylation in iPSCs from SMA1 and SMA2 patients than from healthy individuals. In contrast, the lysosomal trafficking regulator gene is hypermethylated in these cells [109][21] and iPSCs from SMA1 patients and healthy individuals. However, its methylation was significantly higher in iPSCs from SMA2 patients than from SMA1 patients and healthy individuals (Figure 2A).
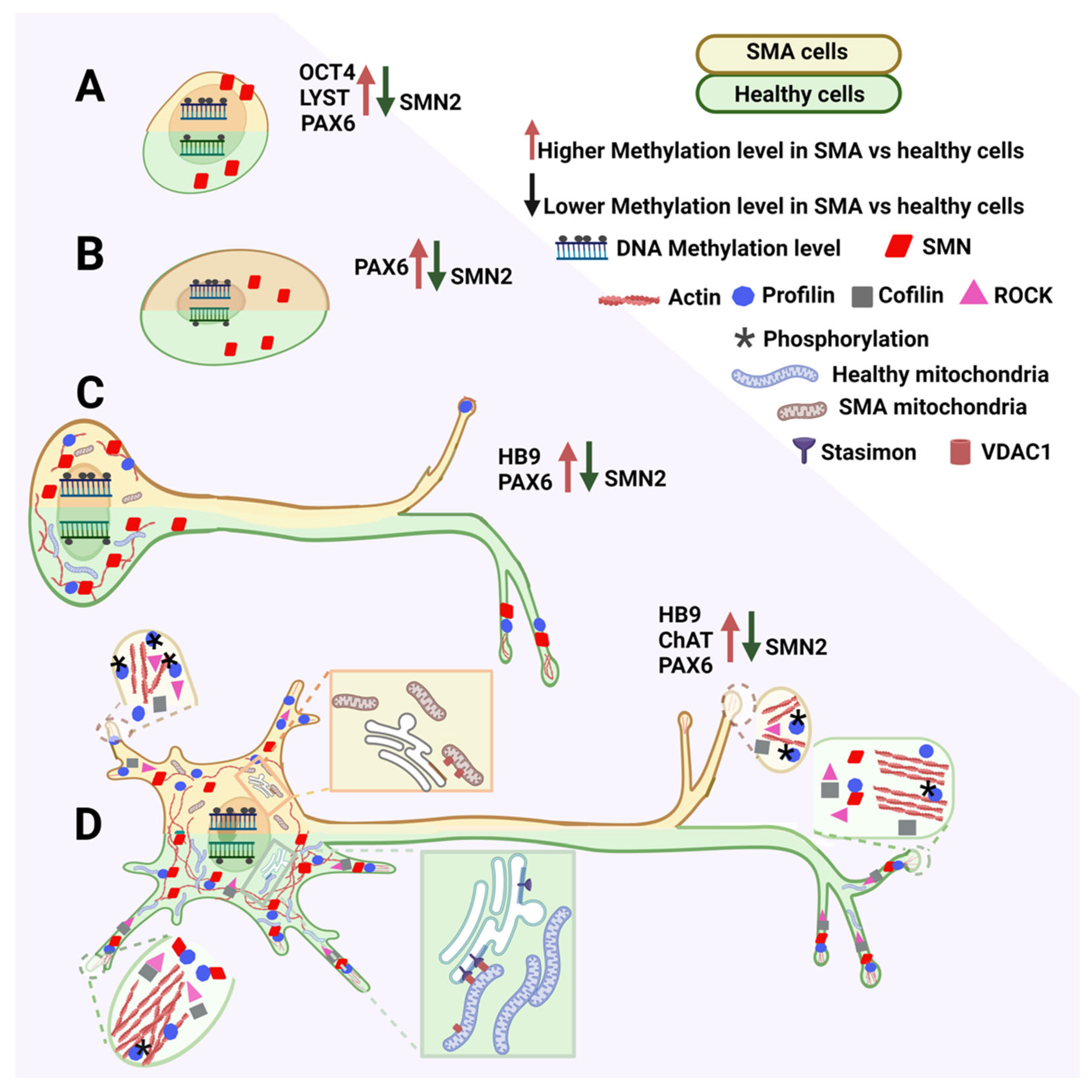
Figure 2. Scheme describing altered interactions in spinal muscular atrophy (SMA) cells during MN development. Each cell drawing is divided in two: the green-shaded half represents cells from healthy individuals, while the orange-shaded half represents cells from SMA patients. Reduced SMA2 methylation is accompanied by an increase in the methylation of various genes. The low SMN protein level alters different pathways and changes protein interactions either at the soma or neurite level. SMA patient mitochondria do not interact properly with the endoplasmic reticulum and have an inappropriate morphology. (A): iPSCs; (B): NEPs; (C): IMNs; and (D): MMNs. OCT4: POU class 5 homeobox 1; LYST: lysosomal trafficking regulator; PAX6: paired box 6; HB9: motor neuron and pancreas homeobox 1 gene; and CHAT: Choline acetyltransferase.
These findings indicate that methylation plays an important role in maintaining iPSC pluripotency. Remarkably, significant increases in OCT4 methylation levels in SMA cells and spalt-like transcription factor 4 gene methylation levels in SMA1 patient-derived cells were present in the three final MN differentiation stages (MN precursor, IMN, and MMN).
Methylation analysis of genes involved in neural differentiation and functioning indicated that primary pathways involved in MN differentiation and maturation are heavily affected in SMA. For example, methylation levels of the MN and pancreas homeobox 1 gene activated during MN maturation [110][22] were inversely correlated with its expression level [111][23]. Its methylation levels were also significantly increased in IMNs and MMNs reprogrammed from SMA1 patient cells, most likely reducing transcription efficiency (Figure 2C,D).
Choline acetyltransferase (CHAT) is a terminal MN marker expressed in MMNs [110][22]. It showed significantly increased methylation in iPSC-derived MNs in cell cultures from a severe SMA case that might affect its expression [106][18] (Figure 2D). While low CHAT levels are correlated with the loss of MN function, decreased CHAT activity in SMA MNs is not the main factor causing their degeneration [71][24]. Instead, acetylcholine receptor clustering changes were described in early SMA1 development [112][25]. Nevertheless, increased CHAT levels might be important in ameliorating MN pathology [113][26].
Plastin 3 (PLS3) and neurocalcin delta (NCALD) are of particular interest since there are many reports assessing their role as SMA modifiers [114,115][27][28]. Indeed, PLS3 acts as a positive disease modifier via endocytosis regulation and is capable of binding to actin and regulating cytoskeletal dynamics through various mechanisms, including actin filament bundling [88,116][29][30]. NCALD adversely affects endocytosis and SMA severity. However, methylation analysis was inconclusive due to the low number of SMA specimens [106][18]. Nevertheless, paired box 6 (Pax6) expression initiates at embryonic day 8 in mice and is present in the neural plate formed by proliferating neuroepithelial cells. Pax6 expression is localized to the spinal cord, forebrain, and hindbrain during regionalized neural tube organization two days later. After birth, Pax6 is strongly expressed in neurons in the amygdala, cerebellum, thalamus, and olfactory bulb. However, it is only modestly expressed in the hippocampus’ subgranular zone (SGZ) and the SVZ [117][31].
In the SGZ, Pax6 is expressed in the SGZ’s neural stem and early progenitor cells. In the SVZ, it persists in migrating immature and mature neurons and is required for the specification of dopaminergic periglomerular cells and GABAergic granule cells. In vitro experiments have shown that PAX6 is more abundant in early differentiation phases, specifically in dorsal fate specification, which is an active proliferating stage [103][15]. Postnatally, the downstream gene, fatty-acid-binding protein 7 (FABP7), has a pivotal role in maintaining neural stem and progenitor cell proliferation during hippocampal neurogenesis. Indeed, in vitro studies also indicated that FABP7 expression is higher in NPCs [103][15].
PAX6 methylation is consistently higher in all differentiation stages in SMA-derived cells. In particular, PAX6 expression increases neurogenesis by human striatal NSCs [118][32]. Therefore, increased methylation and consequent inactivation of PAX6 in NEPs and also iPSCs (Figure 2A) and IMNs and MMNs (Figure 2C,D) [106][18] is associated with reduced stemness in SMA-derived NSCs. Significantly increased PAX6 promoter methylation in cells from SMA1 and SMA2 patients at each consecutive neural differentiation stage might be responsible for changes in PAX6 expression and disrupt the maturation of MNs already damaged in the SMA condition [106][18] (Figure 2).
The methyl CpG-binding protein 2 (MECP2) is disrupted in the Rett syndrome and is a major mCH methyl marker reader. Indeed, it is clear that MECP2 accumulates in neurons throughout postnatal development in parallel with mCH [119][33] reaching an expression level comparable with that of histone H4 [120][34], MECP2 expression in neurons is essential for nervous system function [121][35], and does not change during iPSC differentiation [103][15].
A very recent study found a relationship between MECP2 and SMN2 [122][36]. Indeed, MECP2 binds the SMN2 promoter at nts −167 to −43, and when this site is blocked by antisense oligonucleotides, at −372 to −43, SMN2 expression is additively enhanced with the drug nusinersen [122][36]. This result also translates to an FL-SMN protein increase in patient fibroblasts, an increase in the transactional quadricep and intercostal muscle areas of the mouse model ∆7 (FVB.Cg-Grm7Tg(SMN2)89Ahmb Smn1tm1Msd Tg(SMN2 ∗ delta7) 4299Ahmb/J), and increased survival [122][36].
Conversely, the Tudor domain is involved in the methylation reading of proteins, particularly histones. Indeed, the Tudor domain of Tudor-domain-containing protein 3 recognizes arginine demethylation with asymmetric H4R3, H3R17, and H3R2 dimethylation. It was discovered over two decades ago that SMN binds dimethylated glycine and arginine-rich motifs of snRNP Sm’s D1 and D3 via its Tudor domain [123,124][37][38].
2. SMN Protein Dosage and NSCs Properties
A milestone study [125][39], produced NSCs from embryonic day 14.5 mouse embryo striata from a severe SMA model with Smn1/2 genotypes. These cells showed proliferation and clonal capabilities similar to those obtained from wildtype animals. However, they produced fewer Tuj1-positive neuronal cells, with fewer and shorter neurites. Interestingly, the reduction in Tuj1-positive cells was related to an increase in nestin-positive cells, indicating that these cells have some differentiation impairment.
The reduction in neurite numbers and lengths in SMA-derived neuronal cells can be directly related to alterations in various pathways in which SMN is involved. For example, profilin proteins can bind concurrently to actin (Figure 2C,D) and proteins containing the PLP domain [126][40] which is also present in SMN and encoded by exon 5 [127][41]. In mammals, the 12–15 kDa profilin proteins are encoded by four genes. Profilin-1 is ubiquitously expressed, while profilin-2′s most abundant splice variant, isoform 2a, is predominantly present in the nervous system [128][42]. Profilins exert two main effects on F-actin formation. They bind to actin monomers and ADP-ATP exchange for G-actin, inhibiting F-actin formation [129][43]. Moreover, profilin-2 has a greater affinity for PLP domains than profilin-1 [130][44]. Indeed, the SMN-profilin-2 interaction is more noticeable than the SMN-profilin-1 interaction. Profilin-2′s primary expression in nervous tissue explains why SMA shows neuron degeneration but no actin dynamic alteration in other cells [56,127][41][45] (Figure 2C,D). Rho-associated kinase (ROCK) regulates profilin-2a via phosphorylation [131][46] resulting in its hyperphosphorylation. This mechanism contrasts with other ROCK targets, such as cofilin, which are hypophosphorylated (Figure 2D) [127][41].
Reduced neurite length was also found using iPSCs from healthy individuals or SMA1 or SMA3 patients [132][47]. After iPSCs creation, these cells were differentiated into NSCs and then MNs. Interestingly, the average total neurite length of both SMA clones and neurite projection from SMA3 clusters were much longer than that of the SMA1 clusters [132][47].
Wildtype animals had lower Smn1 expression in the thalamic region than in the hippocampus. Similarly, this region’s cell density, morphology, and proliferation are strongly affected in SMA animals. Indeed, morphological assessment of an Smn2/2;SMN2 mouse model of severe SMA at pre- and late-symptomatic time points found hippocampal-specific effects from reduced SMN levels. In particular, the hippocampal dentate gyrus was noticeably smaller in Smn2/2;SMN2 mice at late symptomatic time points [135][48].
3. SMA, Mitochondria and NSCs
Unlike mature neurons, NSCs use glycolysis and the pentose phosphate pathway for their cellular metabolism, producing energy (ATP) and metabolites (pyruvate and nicotinamide adenine dinucleotide phosphate) that are essential building blocks for aa and nt synthesis to support their high cell division rates [136][49].
NSCs derived from mouse cortex [137,138][50][51], human ESCs (hESCs) [139][52], adult NPCs derived from human iPSCs [140][53], and adult mouse NPCs [141][54].
NSC proliferation and neuronal cell differentiation are accompanied by morphological or functional modifications at the mitochondrial level. These changes are true during embryonic and adult neurogenesis and NSC proliferation and differentiation into NPCs that differentiate into mature neurons. Since mature neurons need more energy to sustain homeostasis and support their specialized functions than stem cells, switching from glycolysis to oxidative phosphorylation is necessary during neuronal differentiation [142][55].
These changes follow a progression in mitochondrial morphology, varying from fragmented to elongated throughout the differentiation stages. The currently accepted concept is that before differentiation, mitochondria are poorly functional with a fragmented morphology and immature cristae structure in early development phases [143,144,145][56][57][58] or different tissues’ adult stem cells [146][59]. They become activated during differentiation through mitochondrial elongation and increased cristae number [147][60]. This aspect was also shown in NSCs during embryonic and adult neurogenesis, with a change in mitochondrial morphology as NSCs began to differentiate first to a neuronal fate, as NPCs, and then into neurons [142][55].
It is interesting to consider the effects of SMN deficiency or absence in the mitochondria of SMA NSCs in the context of these hypotheses. Contrasting with earlier findings [148[61][62],149], it appears that SMN directly or indirectly interacts with many mitochondrial proteins. SMN directly interacts with mitochondrial proteins such as translocases located on the inner membrane, such as translocase of inner mitochondrial membrane 50 [150][63], p53, and B cell lymphoma-2. SMN also has indirect interactions via SMN-interacting proteins that localize to mitochondria, such as NCALD and stasimon, which interact with voltage-dependent anion-selective channel 1 (VDAC1) between the endoplasmic reticulum and mitochondrial membranes [151][64] (Figure 2D). The VDAC1-stasimon interaction could be disrupted by reduced SMN levels, this effect links mitochondrial protein import with SMA pathology [152][65] (Figure 2D).
These aspects correlate with mitochondrial morphology and size. However, other cell types are also associated with the mitochondrion’s energetic and metabolic output. Mitochondrial morphological abnormalities are present in animal SMA models, such as alterations in cristae and larger and smaller sizes [153][66] (Figure 2C,D).
Additionally, the knockdown of Smn in a cellular model (NSC-34, originally produced by fusing MN-rich mouse embryonic spinal cord cells with neuroblastoma N18TG2 cells as undifferentiated cells similar to NSCs) induced many changes in mitochondrial properties [149][62]. In the initial Smn knockdown phase, mitochondrial activity increased to compensate for the energy deficit, suggesting this energy demand can be overcome in early disease phases. Nevertheless, when energy demand increases, such as during MN maturation [149][62], SMN protein levels are insufficient. Indeed, mitochondria from iPSCs-derived MNs from SMA1 patients showed reduced trafficking, number, membrane potential, and size [154][67].
Therefore, it is possible that NSCs obtained from neurogenic brain areas of SMA animal models or humans or produced from iPSCs behave similarly to healthy NSCs since energy requirements at this development stage can be met without mitochondria.
References
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402.
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257.
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220.
- Stec, I.; Nagl, S.B.; van Ommen, G.J.; den Dunnen, J.T. The PWWP domain: A potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 2000, 473, 1–5.
- Qiu, C.; Sawada, K.; Zhang, X.; Cheng, X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 2002, 9, 217–224.
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222.
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905.
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93.
- Clemens, A.W.; Gabel, H.W. Emerging Insights into the Distinctive Neuronal Methylome. Trends Genet. 2020, 36, 816–832.
- Clemens, A.W.; Wu, D.Y.; Moore, J.R.; Christian, D.L.; Zhao, G.; Gabel, H.W. MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol. Cell 2020, 77, 279–293.e278.
- Stroud, H.; Su, S.C.; Hrvatin, S.; Greben, A.W.; Renthal, W.; Boxer, L.D.; Nagy, M.A.; Hochbaum, D.R.; Kinde, B.; Gabel, H.W.; et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 2017, 171, 1151–1164.e1116.
- Kozlenkov, A.; Li, J.; Apontes, P.; Hurd, Y.L.; Byne, W.M.; Koonin, E.V.; Wegner, M.; Mukamel, E.A.; Dracheva, S. A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv. 2018, 4, eaau6190.
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384.
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017, 357, 600–604.
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.K.; Micali, N.; Wang, Y.; Colantuoni, C.; Straub, R.E.; Hoeppner, D.J.; et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11, 462.
- Hauke, J.; Riessland, M.; Lunke, S.; Eyüpoglu, I.Y.; Blümcke, I.; El-Osta, A.; Wirth, B.; Hahnen, E. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 2009, 18, 304–317.
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537.
- Maretina, M.A.; Valetdinova, K.R.; Tsyganova, N.A.; Egorova, A.A.; Ovechkina, V.S.; Schiöth, H.B.; Zakian, S.M.; Baranov, V.S.; Kiselev, A.V. Identification of specific gene methylation patterns during motor neuron differentiation from spinal muscular atrophy patient-derived iPSC. Gene 2022, 811, 146109.
- Hattori, N.; Nishino, K.; Ko, Y.G.; Ohgane, J.; Tanaka, S.; Shiota, K. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J. Biol. Chem. 2004, 279, 17063–17069.
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 2010, 463, 1042–1047.
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7, e1002085.
- Du, Z.W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.L.; Zhong, X.; Fan, F.; Zhang, S.C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626.
- Ferguson, S.; Gautrey, H.E.; Strathdee, G. The dual role of HLXB9 in leukemia. Pediatr. Blood Cancer 2011, 56, 349–352.
- Soler-Botija, C.; Cuscó, I.; Caselles, L.; López, E.; Baiget, M.; Tizzano, E.F. Implication of fetal SMN2 expression in type I SMA pathogenesis: Protection or pathological gain of function? J. Neuropathol. Exp. Neurol. 2005, 64, 215–223.
- Yoshida, H.; Yabuno, A.; Fujiwara, K. Critical appraisal of bevacizumab in the treatment of ovarian cancer. Drug Des. Devel. Ther. 2015, 9, 2351–2358.
- Kim, J.; Shin, K.; Cha, Y.; Ban, Y.H.; Park, S.K.; Jeong, H.S.; Park, D.; Choi, E.K.; Kim, Y.B. Neuroprotective effects of human neural stem cells over-expressing choline acetyltransferase in a middle cerebral artery occlusion model. J. Chem. Neuroanat. 2020, 103, 101730.
- Podgurskaya, A.D.; Tsvelaya, V.A.; Slotvitsky, M.M.; Dementyeva, E.V.; Valetdinova, K.R.; Agladze, K.I. The Use of iPSC-Derived Cardiomyocytes and Optical Mapping for Erythromycin Arrhythmogenicity Testing. Cardiovasc. Toxicol. 2019, 19, 518–528.
- Farooq, F.; Abadía-Molina, F.; MacKenzie, D.; Hadwen, J.; Shamim, F.; O’Reilly, S.; Holcik, M.; MacKenzie, A. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum. Mol. Genet. 2013, 22, 3415–3424.
- Giganti, A.; Plastino, J.; Janji, B.; Van Troys, M.; Lentz, D.; Ampe, C.; Sykes, C.; Friederich, E. Actin-filament cross-linking protein T-plastin increases Arp2/3-mediated actin-based movement. J. Cell Sci. 2005, 118, 1255–1265.
- Delanote, V.; Vandekerckhove, J.; Gettemans, J. Plastins: Versatile modulators of actin organization in (patho) physiological cellular processes. Acta Pharmacol. Sin. 2005, 26, 769–779.
- Osumi, N.; Shinohara, H.; Numayama-Tsuruta, K.; Maekawa, M. Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells 2008, 26, 1663–1672.
- Kallur, T.; Gisler, R.; Lindvall, O.; Kokaia, Z. Pax6 promotes neurogenesis in human neural stem cells. Mol. Cell Neurosci. 2008, 38, 616–628.
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell Neurosci. 2004, 27, 306–321.
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468.
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2019, 147, 1181–1194.
- Wang, J.; Bai, J.; OuYang, S.; Wang, H.; Jin, Y.; Peng, X.; Ge, X.; Jiao, H.; Zou, J.; He, C.; et al. Antisense oligonucleotides targeting the SMN2 promoter region enhance SMN2 expression in spinal muscular atrophy cell lines and mouse model. Hum. Mol. Genet. 2022, 31, 1635–1650.
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117.
- Sprangers, R.; Groves, M.R.; Sinning, I.; Sattler, M. High-resolution X-ray and NMR structures of the SMN Tudor domain: Conformational variation in the binding site for symmetrically dimethylated arginine residues. J. Mol. Biol. 2003, 327, 507–520.
- Shafey, D.; MacKenzie, A.E.; Kothary, R. Neurodevelopmental abnormalities in neurosphere-derived neural stem cells from SMN-depleted mice. J. Neurosci. Res. 2008, 86, 2839–2847.
- Kaiser, D.A.; Goldschmidt-Clermont, P.J.; Levine, B.A.; Pollard, T.D. Characterization of renatured profilin purified by urea elution from poly-L-proline agarose columns. Cell Motil. Cytoskeleton. 1989, 14, 251–262.
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tönges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 2011, 20, 4865–4878.
- Jockusch, B.M.; Murk, K.; Rothkegel, M. The profile of profilins. Rev. Physiol. Biochem. Pharmacol. 2007, 159, 131–149.
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72.
- Nodelman, I.M.; Bowman, G.D.; Lindberg, U.; Schutt, C.E. X-ray structure determination of human profilin II: A comparative structural analysis of human profilins. J. Mol. Biol. 1999, 294, 1271–1285.
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527.
- Da Silva, J.S.; Medina, M.; Zuliani, C.; Di Nardo, A.; Witke, W.; Dotti, C.G. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J. Cell Biol. 2003, 162, 1267–1279.
- Lin, X.; Li, J.J.; Qian, W.J.; Zhang, Q.J.; Wang, Z.F.; Lu, Y.Q.; Dong, E.L.; He, J.; Wang, N.; Ma, L.X.; et al. Modeling the differential phenotypes of spinal muscular atrophy with high-yield generation of motor neurons from human induced pluripotent stem cells. Oncotarget 2017, 8, 42030–42042.
- Wishart, T.M.; Huang, J.P.; Murray, L.M.; Lamont, D.J.; Mutsaers, C.A.; Ross, J.; Geldsetzer, P.; Ansorge, O.; Talbot, K.; Parson, S.H.; et al. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet. 2010, 19, 4216–4228.
- Fawal, M.A.; Davy, A. Impact of Metabolic Pathways and Epigenetics on Neural Stem Cells. Epigenet Insights 2018, 11, 2516865718820946.
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247.
- Knobloch, M.; Jessberger, S. Metabolism and neurogenesis. Curr. Opin. Neurobiol. 2017, 42, 45–52.
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P. Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev. 2015, 24, 1984–1994.
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674.e659.
- Knobloch, M.; Braun, S.M.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, O.; Suter, U.; Machado, R.A.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230.
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 1518.
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sánchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417.
- Facucho-Oliveira, J.M.; Alderson, J.; Spikings, E.C.; Egginton, S.; St John, J.C. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J. Cell Sci. 2007, 120, 4025–4034.
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733.
- Khacho, M.; Slack, R.S. Mitochondrial dynamics in the regulation of neurogenesis: From development to the adult brain. Dev. Dyn. 2018, 247, 47–53.
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873.
- Jablonka, S.; Bandilla, M.; Wiese, S.; Bühler, D.; Wirth, B.; Sendtner, M.; Fischer, U. Co-regulation of survival of motor neuron (SMN) protein and its interactor SIP1 during development and in spinal muscular atrophy. Hum. Mol. Genet. 2001, 10, 497–505.
- Acsadi, G.; Lee, I.; Li, X.; Khaidakov, M.; Pecinova, A.; Parker, G.C.; Hüttemann, M. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J. Neurosci. Res. 2009, 87, 2748–2756.
- Xu, H.; Somers, Z.B.; Robinson, M.L.; Hebert, M.D. Tim50a, a nuclear isoform of the mitochondrial Tim50, interacts with proteins involved in snRNP biogenesis. BMC Cell Biol. 2005, 6, 29.
- Van Alstyne, M.; Lotti, F.; Dal Mas, A.; Area-Gomez, E.; Pellizzoni, L. Stasimon/Tmem41b localizes to mitochondria-associated ER membranes and is essential for mouse embryonic development. Biochem. Biophys. Res. Commun. 2018, 506, 463–470.
- James, R.; Chaytow, H.; Ledahawsky, L.M.; Gillingwater, T.H. Revisiting the role of mitochondria in spinal muscular atrophy. Cell Mol. Life Sci. 2021, 78, 4785–4804.
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406.
- Xu, C.C.; Denton, K.R.; Wang, Z.B.; Zhang, X.; Li, X.J. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis. Model. Mech. 2016, 9, 39–49.
More