The tumor suppressor p53 is a transcription factor that regulates the expression of dozens of target genes and diverse physiological processes. To precisely regulate the p53 network, p53 undergoes various post-translational modifications and alters the selectivity of target genes. Acetylation plays an essential role in cell fate determination through the activation of p53. Ap53 acts as a transcriptionalthough the acetylation of p53 has been examined, the underlying regulatory mechanisms remain unclear and, thus, have attracted the interest of researcher factor that regulates more than 200 target genes and exerts a number of biological effects. Post-translational modifications play an essential role in the precise regulation of the selectivity of different target genes and subsequent physiological outcomes.
- p53
- post-translational modification
- acetylation
- deacetylation
1. p53: Guardian of the Genome
2. p53 Acetylation
p53 is a nuclear protein that comprises 393 amino acids and has the domain structure shown in Figure 1. It contains an N-terminal transactivation domain, proline-rich domain, a centrally located sequence-specific DNA-binding domain (DBD), and a C-terminal tetramerization domain followed by the extreme C-terminal domain (CTD). p53 was the first non-histone protein reported to be acetylated [9]. The acetylation site on p53 has been detected in the following three regions: CTD, DBD, and between these two domains.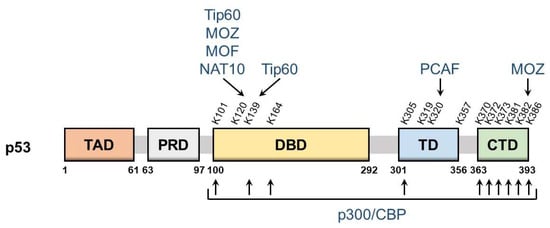
3. Regulators of p53 Acetyltransferase
As shown in Figure 2, multiple factors are involved in the modulation of p53 acetyltransferases. WResearchers herein list regulators that affect the p53 network through their effects on HAT activity.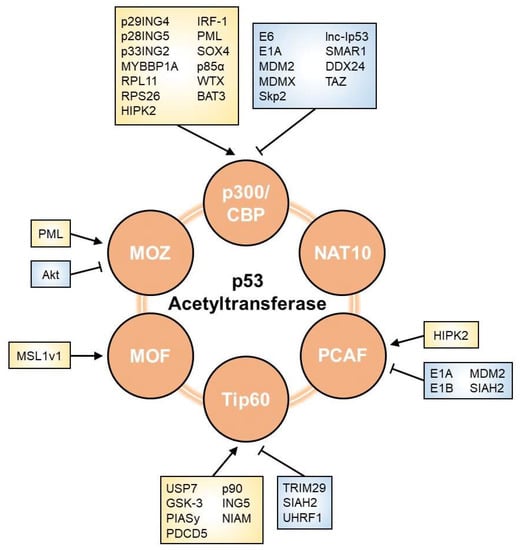
3.1. p300/CBP
Various regulatory modes of p300/CBP activity towards p53 have been reported. Several viral proteins are known to regulate p300/CBP. The E6 protein is one of the oncoproteins encoded by oncogenic human papillomavirus 16 and 18 [64][18]. E6 stimulates the degradation of p53 via a well-known mechanism in which E6 interacts with p53 and promotes its degradation via the ubiquitin–proteasome system. E6 also binds to p300 and CBP and inhibits their co-activation, which blocks p300/CBP-mediated p53 acetylation and down-regulates p53 transcriptional activity [65,66][19][20]. E1A inhibits p300-mediated p53 acetylation [67][21]. It also directly interacts with the HAT domain of p300 and suppresses its HAT activity. In addition, E1A reportedly reduced the p53-dependent transactivation of p21 and Bcl-2-associated X (BAX), leading to the disruption of p53-mediated cell cycle arrest and apoptosis [35][22]. Some proteins related to E3 ubiquitin ligases have been suggested to inhibit p300/CBP activity. MDM2 directly binds to p300/CBP and suppresses its acetyltransferase activity. It was also shown to attenuate p53 acetylation by p300/CBP, which impaired p53 transcriptional activity [68,69][23][24]. MDMX, a homolog of MDM2, was found to inhibit the acetylation of p53 by p300/CBP [70][25]. S phase kinase-associated protein 2 (Skp2), a substrate recognition factor in the SKP-cullin-F-box (SCF) ubiquitin ligase complex, inhibits p300 and suppresses the transcriptional activity of p53 [71][26]. Skp2 directly binds to p300 via the cysteine–histidine-rich regions, CH1 and CH3, which are also p53-binding regions. As Skp2 antagonizes p300-p53 binding, it represses p300-mediated p53 acetylation at K382 as well as the p53-dependent transactivation of both cell cycle arrest genes and pro-apoptotic genes. Some members of the inhibitor of growth proteins (ING) family have also been shown to modulate p300/CBP. p29ING4 and p28ING5 both interact with p300 and promote p53 acetylation at K382, resulting in the p53-mediated transactivation of the p21 gene [72][27]. p33ING2 and p300 form a complex with p53, which enhances p53 acetylation at K382 [73,74][28][29]. p33ING2 suppresses cell proliferation through the activation of p53 via p300-mediated acetylation. Multiple nucleolar proteins, including Myb-binding protein 1a (MYBBP1A), ribosomal protein L11 (RPL11), and ribosomal protein S26 (RPS26), are involved in the regulation of p300/CBP. MYBBP1A directly binds to p53 and strengthens the p53-p300 interaction to promote p53 acetylation in response to ribosomal stress, thereby enhancing p53-mediated transcription [75][30]. RPL11 plays a crucial role in the enhancement of p53-p300 binding and subsequent p300-mediated p53 acetylation at K382 upon nucleolar stress [76][31]. RPS26 forms a complex with p53 and p300 in response to DNA damage and may facilitate p300-mediated p53 acetylation [77][32]. The involvement of long non-coding RNA (lncRNA) in the regulation of p300/CBP has been suggested. lncRNA induced by p53 (lnc-Ip53) interacts with p300 and suppresses p300 activity, leading to the abrogation of p53 acetylation [78][33]. Since lnc-Ip53 is a direct transcriptional target of p53, p53/lnc-Ip53/p300 may form a negative feedback loop in the regulation of p53 activity. Other proteins are also known to positively regulate p300/CBP. Homeodomain interacting protein-kinase 2 (HIPK2), a nuclear serine/threonine kinase, directly binds to p300 and phosphorylates p300, which enhances its HAT activity [79][34]. Moreover, HIPK2 promotes p300-mediated p53 acetylation at K382 and influences the selective transactivation of pro-apoptotic p53 target genes [80,81][35][36]. To trigger the HIPK2-mediated induction of p53 pro-apoptotic genes, p53 needs to be modified by both S46 phosphorylation and K382 acetylation. Interferon regulatory factor 1 (IRF-1) binds to p300 and strengthens the interaction between p300 and the LXXLL transactivation domain of p53 [82][37]. IRF-1 enhances p300-mediated p53 acetylation at K373 and K382, and stimulates the transactivation of p21. Promyelocytic leukemia (PML), a nuclear protein, forms a complex with p53-CBP and promotes p53 acetylation at K382 [83][38]. Moreover, PML inhibits the SCFFbx3-mediated degradation of p300 [84][39]. The PML protein localizes to discrete nuclear speckles called PML nuclear bodies (PML-NBs) [85][40]. When p300 is located within PML-NBs, it is stabilized by PML. However, when p300 is located outside of PML-NBs, it is degraded via the ubiquitin–proteasome pathway. Sex Determining Region Y-Box Transcription Factor 4 (SOX4), a transcriptional factor involved in the regulation of embryonic development and cell fate determination, enhances p53 acetylation at K373 and K382 by interacting with p300/CBP and promoting the formation of the p300/CBP/p53 complex [86][41]. SOX4 may also interact with p53, which inhibits MDM2-dependent p53 ubiquitination and degradation. p85α, a regulatory subunit of phosphatidylinositol-3-kinase, interacts with p300 and augments p300-p53 binding, which leads to p53 transactivation under UVB exposure [87][42]. p85α is required for the induction of mouse p53 acetylation at K370, followed by K373 of human p53. Wilms tumor gene on X chromosome (WTX), which is frequently inactivated in Wilms tumors, augments p53-CBP binding, thereby promoting CBP-mediated p53 acetylation at K373 and K382 and its transcriptional activation [88][43]. HLA-B-associated transcript 3 (BAT3), a nucleo-cytoplasmic shuttling protein, regulates the p300-mediated acetylation of p53 during autophagy [89][44]. During starvation, BAT3 facilitates the nuclear translocation of p300 and promotes p300-dependent p53 acetylation at K373, which leads to the transactivation of pro-autophagic p53 target genes such as SESTRIN1. Several proteins have been reported to function as negative regulators of p300/CBP. Scaffold/matrix attachment region-binding protein 1 (SMAR1), a chromatin remodeling protein, was found to repress p300 expression and inhibit p53 acetylation [90][45]. SMAR1 may associate with the p53-p300 complex and antagonize the interaction of p300 with p53. DEAD (Asp-Glu-Ala-Asp) box RNA helicase 24 (DDX24), a family member of DEAD-box RNA helicases, negatively regulated the transcriptional ability of p53 by suppressing the HAT activity of p300 [91][46]. DDX24 competes with p53 in its interaction with p300 and inhibits the p300-mediated acetylation of p53 at K164 and K382. DDX24 suppresses p53-dependent cell cycle arrest and senescence. Transcriptional coactivator with a PDZ-binding motif (TAZ), a transcriptional coactivator of the Hippo pathway, inhibits p300-mediated p53 acetylation at K382, resulting in the attenuation of p53 transcriptional activity [92][47]. TAZ also interacts with p53 to inhibit the p53-p300 interaction and subsequent p53 acetylation. The knockdown of TAZ has been shown to promote p53-mediated cellular senescence.3.2. PCAF
Multiple viral oncoproteins have been implicated in the regulation of PCAF and the subsequent inactivation of p53. E1A directly binds to PCAF via its HAT domain and diminishes its acetyltransferase activity towards p53 [67][21]. E1A was previously shown to repress p53-induced cell cycle arrest and apoptosis [35][22]. Furthermore, E1B interacted with PCAF and competed for PCAF-p53 binding [93][48]. It also inhibited PCAF-mediated p53 acetylation both in vitro and in vivo and reduced the sequence-specific DNA-binding activity of p53. Some E3 ubiquitin ligases also contribute to the suppression of PCAF. MDM2 was previously shown to inhibit PCAF-mediated p53 acetylation at K320 [94,95][49][50]. MDM2 directly bound to PCAF and promoted its degradation via the ubiquitin–proteasome pathway, which repressed the PCAF-mediated transactivation of p53 target genes. Seven in absentia homolog 2 (SIAH2), which is a member of the SIAH E3 ubiquitin ligases family, also reportedly binds to PCAF and promotes its degradation via the ubiquitin–proteasome pathway [96][51]. The ubiquitination ability of SIAH2 is needed for the attenuation of p53 acetylation and its transcriptional activity. Since SIAH2 is p53 target genes, p53 and SIAH2 may form a feedback loop for the regulation of p53 activity [97][52]. As an example of a positive regulator of PCAF, HIPK2 cooperates with PCAF and drives p53 to selectively transactivate the p21 gene [98][53]. Under apoptotic conditions, HIPK2 is activated in response to severe DNA damage and directly phosphorylates p53 at S46, which induces the expression of pro-apoptotic genes [80,99][35][54]. On the other hand, under non-apoptotic conditions, HIPK2 enhances PCAF-mediated p53 acetylation at K320 by promoting the nuclear localization of PCAF, leading to p53 activation and the selective induction of the p21 gene, but not other pro-apoptotic target genes.3.3. Tip60
p53 acetylation activity of Tip60 is mainly regulated by post-translational modifications, including ubiquitination, phosphorylation, and sumoylation. Regarding ubiquitination, various E3 ubiquitin ligases are involved in the inhibition of Tip60. Tripartite motif 29 (TRIM29) interacts with Tip60 and induces its ubiquitination, resulting in the proteasome-mediated degradation of Tip60 [100][55]. TRIM29 reduces Tip60-mediated acetylation of p53 at K120. SIAH2 binds to Tip60 and promotes its degradation via the ubiquitin–proteasome pathway, which abrogates Tip60-mediated p53 acetylation [96][51]. Ubiquitin-like with PHD and RING finger domains 1 (UHRF1) have also been reported to negatively regulate p53 acetylation activity of Tip60 [101][56]. UHRF1 interacts with Tip60 and promotes its proteasomal degradation. UHRF1 suppresses TIP60-mediated p53 acetylation at K120. Furthermore, the deubiquitinase participates in the regulation of Tip60. Ubiquitin-specific protease 7 (USP7) directly interacts with Tip60 and removes its ubiquitin chains. USP7 enhances Tip60 acetyltransferase activity towards p53 at K120 and promotes p53-mediated pro-apoptotic gene expression [102][57]. Phosphatases act as regulators of Tip60. A previous study demonstrated that glycogen synthase kinase-3 (GSK-3) phosphorylated Tip60 on S86 and enhanced its p53K120 acetyltransferase activity. Moreover, the phosphorylation of S86 promoted the induction of p53-mediated PUMA and apoptosis [103][58]. Sumoylation is associated with Tip60 activity. PIASy, a member of the PIAS family of SUMO E3 ligases, positively regulates Tip60. PIASy induced Tip60 sumoylation and promoted the acetylation of p53 at K120, thereby stimulating p53-mediated apoptosis [104][59]. Other factors related to the regulation of Tip60 are as follows. Programmed cell death 5 (PDCD5), an apoptosis-related protein, interacts with Tip60 and promotes its stabilization. PDCD5 enhances the p53 acetylation activity of Tip60, thereby inducing pro-apoptotic gene expression in a p53-dependent manner [105][60]. Moreover, p90, ING5, and nuclear interactor of ARF and Mdm2 (NIAM) regulate Tip60-mediated p53 acetylation at K120 [106,107,108][61][62][63].3.4. MOZ
The mode of regulation of MOZ activity towards p53 remains unclear. PML directly binds to MOZ, which is then recruited into PML-NBs [51][64]. Under cellular stress conditions, MOZ colocalizes with p53 in PML-NBs and promotes p53 acetylation at K120 and K382. These modifications stimulate the p53-mediated transactivation of p21. In contrast, Akt, a serine/threonine kinase, phosphorylates MOZ at T369 and suppresses the interaction between PML and MOZ [51][64]. The phosphorylation of T369 is essential for the negative regulation of MOZ-mediated p53 acetylation.3.5. MOF
Regarding the p53 acetylation activity of MOF, its regulatory mode has not yet been examined in as much detail as those of other p53 acetyltransferases. Male-specific lethal 1v1 (MSL1v1) may form a complex with MOF and markedly enhance p53-dependent transcriptional activity [109][65]. MOF-MSL1v1 is recruited to the promoter region through interactions with p53 and other cofactors and then acetylates p53 at K120, which enhances the p53-mediated transcription of PUMA and BAX.3.6. NAT10
Limited information is currently available on the regulatory mode of the acetylation activity of NAT10 towards p53. Acetyltransferases generally regulate enzymatic activity by autoacetylation [110][66]. NAT10 also appears to be regulated by autoacetylation [111][67]. Several enzymes have been reported to deacetylate NAT10. For example, SIRT1 deacetylates NAT10 and inhibits its rRNA biogenesis function [112][68]. It currently remains unclear whether SIRT1 affects the p53 acetylation activity of NAT10 and, thus, further studies are needed.References
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606.
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622.
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961.
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950.
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606.
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373.
- Kon, N.; Ou, Y.; Wang, S.J.; Li, H.; Rustgi, A.K.; Gu, W. mTOR inhibition acts as an unexpected checkpoInt. in p53-mediated tumor suppression. Genes Dev. 2021, 35, 59–64.
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839.
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626.
- Chao, C.; Wu, Z.; Mazur, S.J.; Borges, H.; Rossi, M.; Lin, T.; Wang, J.Y.; Anderson, C.W.; Appella, E.; Xu, Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell Biol. 2006, 26, 6859–6869.
- Knights, C.D.; Catania, J.; Di Giovanni, S.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544.
- Wang, Y.H.; Tsay, Y.G.; Tan, B.C.; Lo, W.Y.; Lee, S.C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 2003, 278, 25568–25576.
- Joubel, A.; Chalkley, R.J.; Medzihradszky, K.F.; Hondermarck, H.; Burlingame, A.L. Identification of new p53 acetylation sites in COS-1 cells. Mol. Cell Proteom. 2009, 8, 1167–1173.
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136.
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. Embo J. 1999, 18, 5061–5072.
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219.
- Chakravarti, D.; Ogryzko, V.; Kao, H.Y.; Nash, A.; Chen, H.; Nakatani, Y.; Evans, R.M. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 1999, 96, 393–403.
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827.
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo J. 2001, 20, 1331–1340.
- Kobet, E.; Zeng, X.; Zhu, Y.; Keller, D.; Lu, H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 12547–12552.
- Sabbatini, P.; McCormick, F. MDMX inhibits the p300/CBP-mediated acetylation of p53. DNA Cell Biol. 2002, 21, 519–525.
- Kitagawa, M.; Lee, S.H.; McCormick, F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol. Cell 2008, 29, 217–231.
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378.
- Nagashima, M.; Shiseki, M.; Miura, K.; Hagiwara, K.; Linke, S.P.; Pedeux, R.; Wang, X.W.; Yokota, J.; Riabowol, K.; Harris, C.C. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc. Natl. Acad. Sci. USA 2001, 98, 9671–9676.
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell Biol. 2005, 25, 6639–6648.
- Kuroda, T.; Murayama, A.; Katagiri, N.; Ohta, Y.M.; Fujita, E.; Masumoto, H.; Ema, M.; Takahashi, S.; Kimura, K.; Yanagisawa, J. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. Embo J. 2011, 30, 1054–1066.
- Mahata, B.; Sundqvist, A.; Xirodimas, D.P. Recruitment of RPL11 at promoter sites of p53-regulated genes upon nucleolar stress through NEDD8 and in an Mdm2-dependent manner. Oncogene 2012, 31, 3060–3071.
- Cui, D.; Li, L.; Lou, H.; Sun, H.; Ngai, S.M.; Shao, G.; Tang, J. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014, 33, 2225–2235.
- Zhang, L.Z.; Yang, J.E.; Luo, Y.W.; Liu, F.T.; Yuan, Y.F.; Zhuang, S.M. A p53/lnc-Ip53 Negative Feedback Loop Regulates Tumor Growth and Chemoresistance. Adv. Sci. 2020, 7, 2001364.
- Aikawa, Y.; Nguyen, L.A.; Isono, K.; Takakura, N.; Tagata, Y.; Schmitz, M.L.; Koseki, H.; Kitabayashi, I. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. Embo J. 2006, 25, 3955–3965.
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, ncb715.
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85.
- Dornan, D.; Eckert, M.; Wallace, M.; Shimizu, H.; Ramsay, E.; Hupp, T.R.; Ball, K.L. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol. Cell Biol. 2004, 24, 10083–10098.
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210.
- Shima, Y.; Shima, T.; Chiba, T.; Irimura, T.; Pandolfi, P.P.; Kitabayashi, I. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol. Cell Biol. 2008, 28, 7126–7138.
- Zhong, S.; Salomoni, P.; Pandolfi, P.P. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2000, 2, E85–E90.
- Pan, X.; Zhao, J.; Zhang, W.N.; Li, H.Y.; Mu, R.; Zhou, T.; Zhang, H.Y.; Gong, W.L.; Yu, M.; Man, J.H.; et al. Induction of SOX4 by DNA damage is critical for p53 stabilization and function. Proc. Natl. Acad. Sci. USA 2009, 106, 3788–3793.
- Song, L.; Gao, M.; Dong, W.; Hu, M.; Li, J.; Shi, X.; Hao, Y.; Li, Y.; Huang, C. p85α mediates p53 K370 acetylation by p300 and regulates its promoter-specific transactivity in the cellular UVB response. Oncogene 2011, 30, 1360–1371.
- Kim, W.J.; Rivera, M.N.; Coffman, E.J.; Haber, D.A. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol. Cell 2012, 45, 587–597.
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.S.; Hubberstey, A.V.; Palissot, V.; et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 4115–4120.
- Sinha, S.; Malonia, S.K.; Mittal, S.P.; Mathai, J.; Pal, J.K.; Chattopadhyay, S. Chromatin remodelling protein SMAR1 inhibits p53 dependent transactivation by regulating acetyl transferase p300. Int. J. Biochem. Cell Biol. 2012, 44, 46–52.
- Shi, D.; Dai, C.; Qin, J.; Gu, W. Negative regulation of the p300-p53 interplay by DDX24. Oncogene 2016, 35, 528–536.
- Miyajima, C.; Kawarada, Y.; Inoue, Y.; Suzuki, C.; Mitamura, K.; Morishita, D.; Ohoka, N.; Imamura, T.; Hayashi, H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells 2020, 9, 171.
- Liu, Y.; Colosimo, A.L.; Yang, X.J.; Liao, D. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell Biol. 2000, 20, 5540–5553.
- Jin, Y.; Zeng, S.X.; Lee, H.; Lu, H. MDM2 mediates p300/CREB-binding protein-associated factor ubiquitination and degradation. J. Biol. Chem. 2004, 279, 20035–20043.
- Jin, Y.; Zeng, S.X.; Dai, M.S.; Yang, X.J.; Lu, H. MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J. Biol. Chem. 2002, 277, 30838–30843.
- Grishina, I.; Debus, K.; García-Limones, C.; Schneider, C.; Shresta, A.; García, C.; Calzado, M.A.; Schmitz, M.L. SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim. Biophys. Acta 2012, 1823, 2287–2296.
- Matsuzawa, S.; Takayama, S.; Froesch, B.A.; Zapata, J.M.; Reed, J.C. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: Suppression by BAG-1. Embo J. 1998, 17, 2736–2747.
- Di Stefano, V.; Soddu, S.; Sacchi, A.; D’Orazi, G. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21Waf1 after nonapoptotic DNA damage. Oncogene 2005, 24, 5431–5442.
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19.
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys. Acta 2011, 1813, 1245–1253.
- Dai, C.; Shi, D.; Gu, W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1). J. Biol. Chem. 2013, 288, 19581–19592.
- Dar, A.; Shibata, E.; Dutta, A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell Biol. 2013, 33, 3309–3320.
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596.
- Naidu, S.R.; Lakhter, A.J.; Androphy, E.J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle 2012, 11, 2717–2728.
- Xu, L.; Chen, Y.; Song, Q.; Xu, D.; Wang, Y.; Ma, D. PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia 2009, 11, 345–354.
- Dai, C.; Tang, Y.; Jung, S.Y.; Qin, J.; Aaronson, S.A.; Gu, W. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc. Natl. Acad. Sci. USA 2011, 108, 18937–18942.
- Liu, N.; Wang, J.; Wang, J.; Wang, R.; Liu, Z.; Yu, Y.; Lu, H. ING5 is a Tip60 cofactor that acetylates p53 in response to DNA damage. Cancer Res. 2013, 73, 3749–3760.
- Reed, S.M.; Hagen, J.; Tompkins, V.S.; Thies, K.; Quelle, F.W.; Quelle, D.E. Nuclear interactor of ARF and Mdm2 regulates multiple pathways to activate p53. Cell Cycle 2014, 13, 1288–1298.
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900.
- Li, X.; Wu, L.; Corsa, C.A.; Kunkel, S.; Dou, Y. Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol. Cell 2009, 36, 290–301.
- McCullough, C.E.; Marmorstein, R. Molecular Basis for Histone Acetyltransferase Regulation by Binding Partners, Associated Domains, and Autoacetylation. ACS Chem. Biol. 2016, 11, 632–642.
- Cai, S.; Liu, X.; Zhang, C.; Xing, B.; Du, X. Autoacetylation of NAT10 is critical for its function in rRNA transcription activation. Biochem. Biophys. Res. Commun. 2017, 483, 624–629.
- Liu, X.; Cai, S.; Zhang, C.; Liu, Z.; Luo, J.; Xing, B.; Du, X. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 2018, 46, 9601–9616.