Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Dhanashree Murugan.
Targeted protein degradation is a new aspect in the field of drug discovery. Traditionally, developing an antibiotic includes tedious and expensive processes, such as drug screening, lead optimization, and formulation. Proteolysis-targeting chimeras (PROTACs) are new-generation drugs that use the proteolytic mechanism to selectively degrade and eliminate proteins involved in human diseases. The application of PROTACs is explored immensely in the field of cancer, and various PROTACs are in clinical trials.
- PROTAC
- targeted proteolysis
- anti-bacterial drugs
- anti-viral drugs
1. Introduction
Proteolysis-targeting chimeras (PROTACs) are bifunctional protein degraders that use the E3 ubiquitin ligase pathway for the degradation of the protein of interest. A PROTAC molecule consists of three components: a ligand moiety that targets the protein of interest (POI), another ligand that binds to E3 ligase, and a linker, which bridges between these two ligands [1]. The main function of these ligands is to attract E3 ligase and POI and initiate polyubiquitination for degrading POI via the ubiquitin–proteasome system (UPS) (Figure 1) [2]. The ubiquitin–proteasome system is an essential pathway of every eukaryotic cell for maintaining homeostasis and regulating gene transcription and translation, cell cycle, and apoptosis [3]. In the ubiquitin proteolysis pathway, the ubiquitin molecule binds to the ubiquitin-activating enzyme E1. The E1-bound ubiquitin transfers the ubiquitin to the E2-conjugating enzyme, which is later transferred to E3 ligase, and, finally, ubiquitin binds to POI. These are ATP-driven cascades of reaction where the ubiquitin molecule is transferred from one molecule to another and, finally, to POI. Similarly, several ubiquitin molecules bind to the POI, which signals the proteasome to initiate the degradation of the polyubiquitinated POI. This innate protein degradation pathway is utilized for degrading POI.
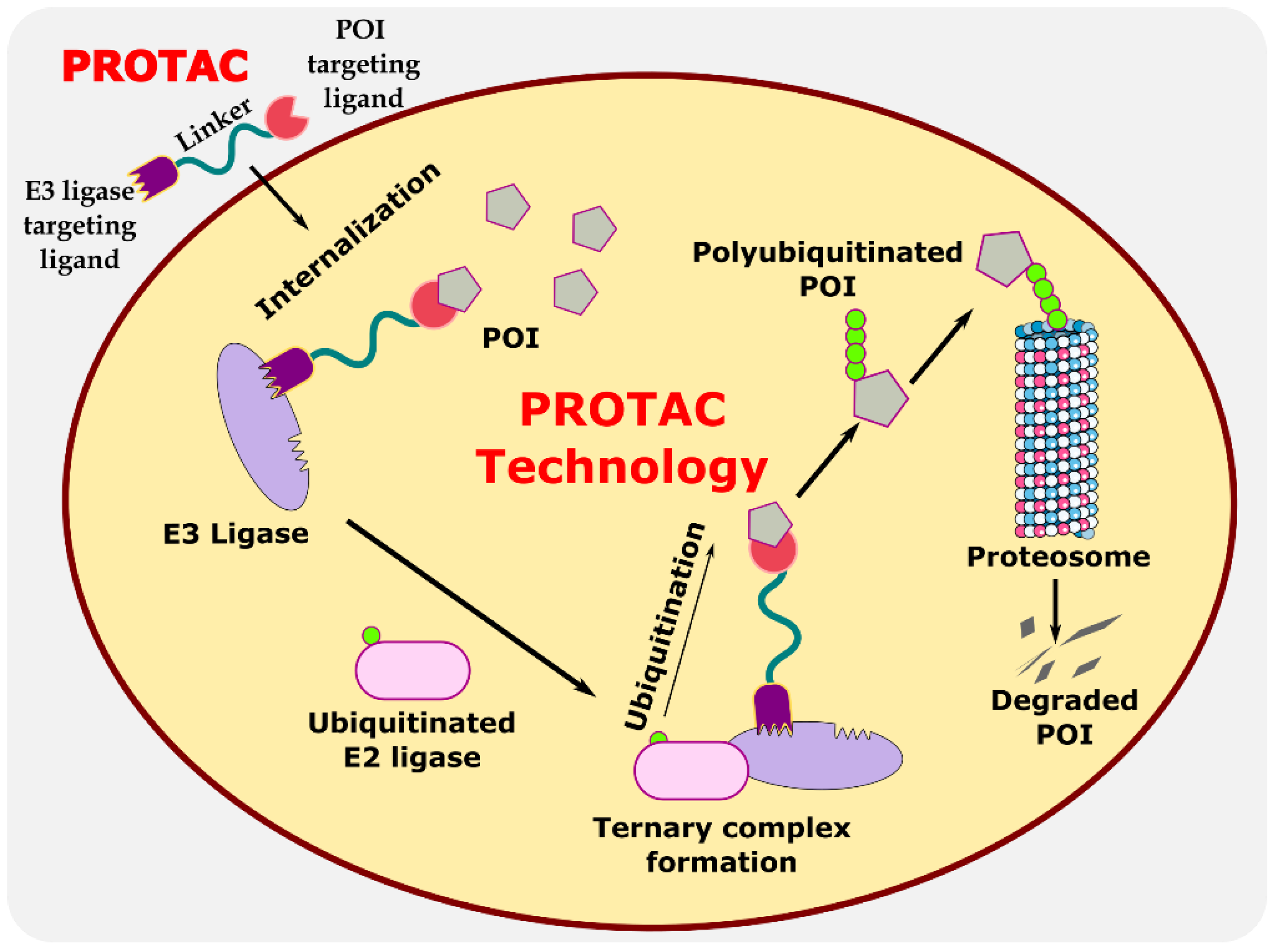
Figure 1.
Illustration explaining the mechanism of PROTAC in targeted protein degradation.
The targeted degradation of the proteins has been explored using various techniques like ligand-induced degradation [4] (LID), hydrophobic tagging (HyT) [5], etc. These techniques later lead to the development of PROTACs [6]. PROTACs are more efficient than conventional small molecule inhibitors [7]. For instance, traditional small molecule inhibitors could only inhibit the activity of certain enzymes or could block the partial function of the protein, while PROTACs can completely eliminate the disease-related proteins [8]. A significantly lower concentration of the drug is required in case of targeted protein degradation using PROTACs as compared to small molecule inhibitors. Many proteins which remain undruggable over the decades, like scaffold proteins, transcriptional factors, or proteins without active binding sites, could be easily targeted by PROTACs and other similar targeted technologies [9]. Such molecules have the great advantages of high selectivity, catalytic, and drugging the undruggable targets.
The first PROTAC molecule was successfully developed in 2001, and, to date, more than 3270 PROTACs have been developed [10]. Some of the PROTAC molecules are currently in different phases of clinical trials, and their initial results have provided a great modality for PROTAC-based degraders (Table 1) [11]. Thus, PROTACs have grabbed the attention of various pharmaceutical companies. Companies such as Arvinas [12], Pfizer [13], Accutar Biotech [14], Bristol Myers Squibb [15], Dialectic Therapeutics [16], Foghorn Therapeutics [17], Kymera Therapeutics [18], Nurix Therapeutics [19], C4 Therapeutics [20], and Cullgen [21] have already entered in the race of clinical trials for their respective PROTAC molecules. It has been predicted that within a few years, approximately 15 PROTAC molecules will be in clinics [22]. Due to the inarguable potential of PROTACs in the current era, researchers are exploring the possibilities of developing new protein degraders for various diseases, such as immunological disorders [23], inflammatory disorders [24[24][25][26],25,26], cancer [27[27][28],28], auto-immune diseases [29], neurological diseases [30], bacterial infections [31], and viral infections [32]. It is undeniable to state that PROTAC-based degraders are highly investigated in the field of cancer research, and many protein degraders are in the pipeline for clinical trials. However, the exploration of PROTACs in the field of anti-microbial remains marginal. This entreviewy is an attempt to highlight the state-of-the-art protein-based degraders targeting microorganisms. It also emphasizes PROTACs as an alternative to antibiotics.
Table 1.
Comprehensive representation of clinical cases of PROTACs.
| Sr. No. | Molecule | Route of Delivery (Dose) | Stage of the Trial | No. of Patients | Targeted Disease | Company | Follow Up Period | Clinical Trial No. | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ARV-110 | Oral (Tablets) once or twice daily for 28 day cycles |
Recruiting (Phase II) | 36 | Prostate cancer | Arvinas, USA | 28 days | [48]NCT03888612 | [33] | ||||||
| 2 | ARV-471 | Oral | Recruiting (Phase II) | 36 | Breast cancer | Arvinas, Pfizer, USA | 28 days | NCT04072952 | [34] | ||||||
| 2 | Hepatitis C virus (HCV) | HCV NS3/4A protease | Telaprevir | CRBN ligand |
|
[50] | 3 | AC682 | Oral | Recruiting (Phase I) | 42 | ||||
| 3 | Influenza A virus | Breast cancer | Accutar Biotech, USA | 18 months | Viral endonuclease PA | Asperphenalenone E |
| NCT05080842 | [51][35] | ||||||
| 4 | ARV-766 | Oral | Recruiting (Phase II) | 60 | Prostate cancer | Arvinas, USA | 6 weeks | NCT05067140 | |||||||
| 4 | Influenza A Virus | Influenza hemagglutinin | Pentacyclic triterpenoid | [ | 36 | ] | |||||||||
| VHL and | CRBN ligand |
|
[52] | 5 | CC-94676 | Oral | Recruiting (Phase I) | 40 | Prostate cancer | Celgene, USA | - | NCT04428788 | |||
| 5 | H1N1 influenza virus | Neuraminidase | Oseltamivir | VHL and CRBN ligand | [ |
|
[53] | 37 | ] | ||||||
| 6 | DT2216 | Intravenous administration | Recruiting (Phase I) | 24 | Liquid and solid tumors | Dialectic Therapeutics, USA | |||||||||
| 6 | SARS-CoV-2 virus | The RNA genome of the SARS-CoV-2 virus | RNA attenuator hairpin (AH) | 28 days |
| NCT04886622 |
| [38] | |||||||
| [ | 54 | ] | 7 | FHD-609 | Intravenous administration | Recruiting (Phase I) | 70 | ||||||||
| 7 | SARS-CoV-2 virus | Spike protein of SARS-CoV-2 | Synovial sarcoma | Antisense oligonucleotide |
| Foghorn Therapeutics, USA | 6 weeks | NCT04965753 | [39] | ||||||
| [ | 55 | ] | 8 | KT-474 | Oral | Completed (Phase I) | 124 | Autoimmune diseases (e.g., AD, HS, RA) | Kymera Therapeutics, USA | 28 days | NCT04772885 | [40] | |||
| 9 | KT-413 | Intravenous administration | Recruiting (Phase I) | 80 | Diffuse large B cell lymphoma (MYD88-mutant) | Kymera Therapeutics, USA | 18 months | NCT05233033 | [41] | ||||||
| 10 | KT-333 | Intravenous administration | Recruiting (Phase I) | 80 | Liquid and solid tumors | Kymera Therapeutics | 18 months | NCT05225584 | [41] | ||||||
| 11 | NX-2127 | Oral | Recruiting (Phase I) | 130 | B cell malignancies | Nurix Therapeutics, USA | 6 months | NCT04830137 | [42] | ||||||
| 12 | NX-5948 | Oral | Recruiting (Phase I) | 130 | B cell malignancies and autoimmune diseases | Nurix Therapeutics, USA | 100 days | NCT05131022 | [43] | ||||||
| 13 | CFT8634 | Oral | Recruiting (Phase II) | 110 | Synovial sarcoma | C4 Therapeutics | 90 days | NCT05355753 | [44] | ||||||
| 14 | Protac MyFit | Intravenous administration | Recruiting (Phase I) | 240 | Sensory impairment (SPD) | University of Southern Denmark | 21 days | NCT04173871 | [45] |
In the current scenario, due to the inappropriate/overuse of antibiotics, the use of antibiotics in the agricultural field and feeding livestock has led to the emergence of resistant strains of pathogens, which is a major threat to humankind. Consequently, pharmaceutical industries consider the development of new antibiotic as potentially effective for a shorter duration and also requires hefty investment [46]. Thus, there is a need to develop new strategies for targeting multidrug-resistant pathogens. Thus, PROTAC could be a glimmer of hope for destroying resistant pathogens [31]. Due to the characteristic nature of direct degrading of the disease-related protein or POI instead of inhibiting them could provide enhanced sensitivity towards multidrug resistant pathogens [47]. Since PROTACs utilize a chemical knockdown approach, an innate cellular mechanism, there are likely fewer chances of the generation of spontaneous mutations in the target protein [8]. There are several approaches for degrading the POI, and they are classified based on the type of degradation system used, namely, the eukaryotic system and prokaryotic system.
2. Eukaryotic System
2.1. Anti-Viral PROTACs
2.1.1. Degradation of Viral Protein
In this section, PROTACs that target viral proteins have been enlisted. PROTAC-based protein degraders are highly explored in this section as compared to other strategies of protein degradation. Many viral proteins have been targeted for protein degradation (Table 2). For instance, Montrose et al. developed a peptide-based PROTAC molecule that targets X-protein, which is an essential protein required for the replication of the hepatitis B virus (HBV). It was also found that the presence of X-protein could also induce hepatocellular Carcinoma (HCC). This PROTAC molecule consists of an ODD degrons (oxygen-dependent degradation) domain, an oligomerization domain, and a cell-penetrating peptide. The ODD degrons domain binds to Von Hippel–Lindau (VHL) E3 ligase, the oligomerization domain interacts with the X-protein, and octa-arginine is used as a CPP for to ease cellular entry. In vitro studies verified the ability of peptide-based PROTACs to efficiently degrade X-protein [48]. In another study, instead of the peptide as a ligand for POI, the authors used telaprevir, an anti-viral peptidomimetic protease inhibitor. They developed three different molecules (DGY-03-081 (2), DGY-04-035 (3), and DGY-08-097 (4)) that target the NS3/4A protease of the hepatitis C virus (Figure 2). Lenalidomide, pomalidomide and novel tricyclic imide moiety were used as the ligands for CRBN E3 ligase. The function of NS3/4A serine protease is to cleave viral polyprotein, which acts as an essential step in viral replication [49]. Thus, degradation of NS3/4A protease via PROTAC will inhibit virion formation and multiplication. These compounds were evaluated in Hep C virus-infected HEK293T cells. Interestingly, all three degraders exhibited anti-viral activity and did not show cytotoxicity to the uninfected cells. Compound DGY-08-097 (4) had the highest degradation ability and the least DC50 value (50 nM at 4 h). One of the reasons for the increased affinity might be due to the tricyclic imide moiety in the DGY-08-097 (4) that showed increased affinity towards CRBN E3 ligase [50].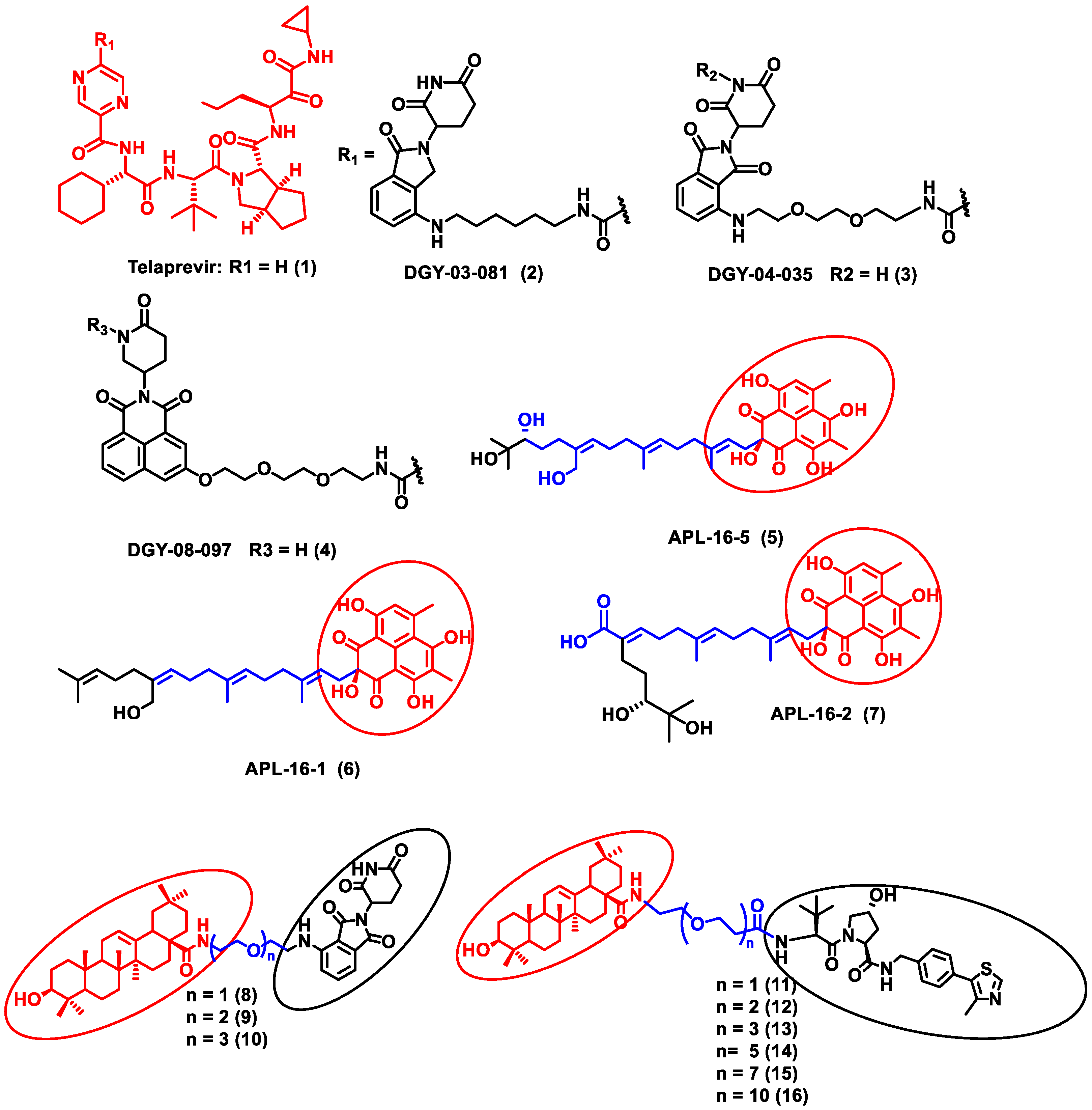
Figure 2. Structures of the PROTAC molecules used for the degradation of viral proteins. The red circle indicates the POI ligand. The blue wavy line indicates the linker, and the black circles indicate the E3 ligand moiety.
Table 2.
Comprehensive information on the targeted degradation of viral protein in the eukaryotic system using PROTAC molecules.
| S. No. | Pathogen | Protein of Interest (POI) | POI Ligand | E3 Ligase Ligand | Research Outcome | Ref. |
|---|---|---|---|---|---|---|
| 1 | Hepatitis B virus (HBV) | X-protein of the hepatitis B virus (HBV) | Oxygen-dependent degradation (ODD) domain of hypoxia-inducible factor (HIF-1a) | VHL ligand |
|
Similarly, the endonuclease polymerase subunit (PA) of influenza virus A was the POI for developing a novel PROTAC molecule. Asperphenalenone E (APL-16-5) (5) was derived from an endophytic fungus Aspergillus, which was used as a ligand for PA. APL-16-5 (5) induces degradation of the viral polymerase subunit (PA) by ubiquitin–proteasome machinery, as it can bind to both the E3 ligase enzyme (TRIM25) and PA. The endonuclease polymerase enzyme is essential in the polymerization of DNA during DNA replication. Derivatives of asperphenalenone, APL-16-1 (6), and APL-16-2 (7) were also synthesized, and the results were compared with the known anti-viral drug ribavirin. HEK293T, A549, and MDCK cells were cultured and infected with influenza virus A WSN/33 for in vitro analysis. The cytotoxicity of APL-16-5 (5) and APL-16-1 (6) against the Influenza virus was in micromolar concentration (EC50) 0.28 to 0.36 μM. Proteosome-mediated degradation of PA with APL-16-5 (5) exhibited a marked decrease in viral RNA components. Later, APL-16-5 (5) was evaluated against influenza virus B, hepatitis C, and Zika viruses. The results from the study confirmed that APL-16-5 is a selective inhibitor for influenza viruses. Dose-dependent studies were conducted to determine the interaction of PA with TRIM25 and concluded that compound 5 induces the destabilization of PA by ubiquitination, and thereby it degrades the PA [51].
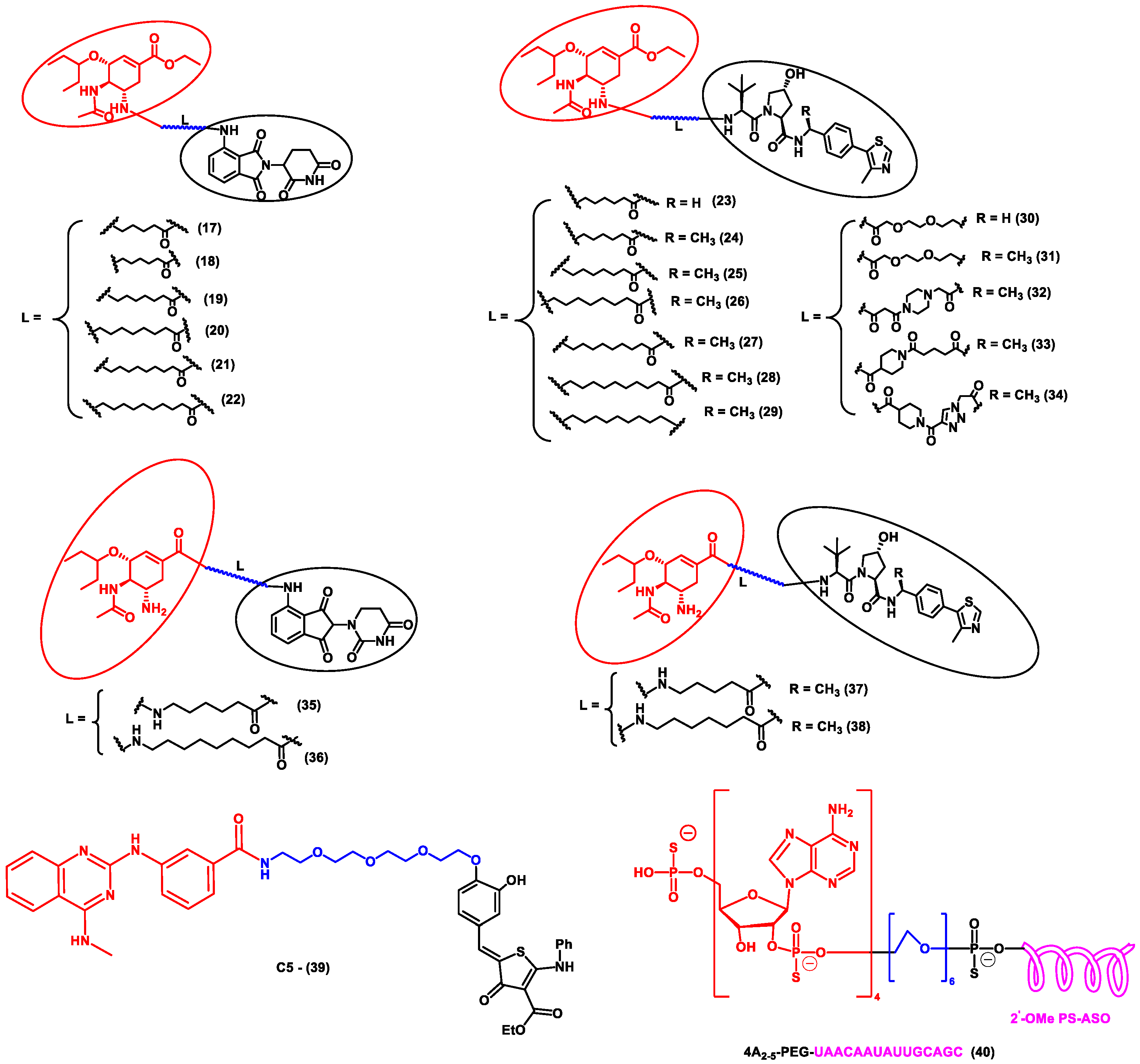
Li et al. designed a pentacyclic triterpenoid group (PTG) containing PROTAC molecule for targeting hemagglutinin (HA) of the influenza virus. Pentacyclic triterpenoids are secondary metabolites present in various medicinal plants, and they possess significant anti-viral activity. Oleanolic acid (OA) and its derivatives are compounds that were selected as the warhead for the PROTAC molecule. OA exhibited anti-viral action against the influenza A/WSN/33 virus, and it had a moderate binding affinity with HA; thus, it became an ideal molecule for PROTAC technology. Two sets of PROTAC molecules (8–10) and (11–16) were designed and studied employing different E3 ligases, such as CRBN and VHL ligands, respectively. HEK293T cells were transfected using HA plasmids, and the level of HA degradation was studied using the synthesized PROTAC molecules. A cell viability assay, immunofluorescence microscopy assay, immunoprecipitation assay, hemagglutination inhibition assay, etc., were performed to evaluate the molecules. Compound 13 (DC50 = 1.44 μM) exhibited the maximum HA depletion as compared to other compounds. This was also validated by molecular docking analysis by Schrodinger Suite. Furthermore, it was concluded from these assays that the VHL ligand containing PROTACs showed better HA degradation [52].
In another independent study conducted by Xu et al., oseltamivir is an approved drug for influenza that targets influenza neuraminidase (NA). Neuraminidase is an essential enzyme for viral replication. They have used oseltamivir-based compounds for targeting neuraminidase and linked them with a discrete variety of E3 ligase ligands such as VHL or CRBN. The amino or carboxylate group of oseltamivir was modified to improve its anti-viral activity. A wide variety of linker combinations like rigid as well as flexible groups like PEG, pyridyl, triazole, and piperazinyl were also involved. A set of PROTAC combinations (17–38) were designed, and from these, N-substituted oseltamivir showed increased potency than the carboxylate-substituted compound. According to the in vitro studies, compound 27 showed the best anti-viral activity having an EC50 of 0.33 µM, which was almost similar to the reference drug oseltamivir phosphate (EC50 = 0.36 µM). Furthermore, interestingly, all the synthesized compounds do not show cytotoxicity towards the normal cells with a concentration up to CC50 > 50 µM. Docking studies indicated that these ternary complexes showed great hydrogen bonding and hydrophobic interactions between neuraminidase and E3 ligase [53]. From these above studies, it could be concluded that there are various strategies being evolved to target viral proteins and inhibit their replication.
Other interesting subcategories of PROTAC are ribonuclease-targeting chimera (RIBOTAC) and nucleic acid-hydrolysis-targeting chimera (NATAC). Both strategies were used to develop novel degraders. In RIBOTAC, RNase is the degrader system, and it degrades viral RNA, while NATAC uses oligonucleotide sequences to identify the POI, and further, they could be degraded by RNase L (specific for ss-RNA). Haniff et al. developed a RIBOTAC degrader that targets the RNA genome of the SARS-CoV-2 virus (Figure 3). RIBOTAC has two major constituents- a small molecule known as C5 (39) and an RNA attenuator hairpin (AH). This RNA attenuator hairpin binds to the RNA genome, and C5 (39) recruits endonucleases present in the cell and initiates the degradation of the viral genome (Figure 4) [54]. This strategy might provide solutions for various viral infections, and the only challenge is identifying and optimizing the appropriate attenuator sequence, which could bind toward a target of interest.
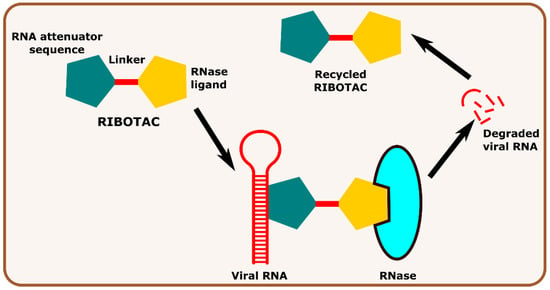
Figure 3.
Schematic representation of the mechanism of action of RIBOTAC molecules in targeting viral RNA.
Figure 4. Structures of the PROTAC molecules are used for the degradation of viral proteins. The red circle indicates the POI ligand; the blue wavy line indicates the linker; the black circles indicate the E3 ligand moiety.
References
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574.
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240.
- Vijayakumar, A.K.; Kuppuswamy, V.; Chinnakkannu, P. Administration of Grape Seed Extract Alleviates Age-Associated Decline in Ubiquitin-Proteasome System and Cardiomyocyte Apoptosis in Rats. Adv. Biol. Chem. 2013, 3, 253–263.
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-Induced Genetic Degradation as a Tool for Target Validation. Drug Discov. Today Technol. 2019, 31, 91–98.
- Choi, S.R.; Wang, H.M.; Shin, M.H.; Lim, H.-S. Hydrophobic Tagging-Mediated Degradation of Transcription Coactivator SRC-1. Int. J. Mol. Sci. 2021, 22, 6407.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorganic Med. Chem. Lett. 2008, 18, 5904–5908.
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Sig. Transduct. Target 2019, 4, 1–33.
- Mayor-Ruiz, C.; Winter, G.E. Identification and Characterization of Cancer Vulnerabilities via Targeted Protein Degradation. Drug Discov. Today Technol. 2019, 31, 81–90.
- Protacdb Server. Available online: http://cadd.zju.edu.cn/protacdb/ (accessed on 11 November 2022).
- Alabi, S.B.; Crews, C.M. Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647.
- Protein Degradation with PROTAC Protein Degraders. Available online: https://www.arvinas.com/ (accessed on 2 November 2022).
- Arvinas and Pfizer Announce Global Collaboration to Develop and Commercialize PROTAC® Protein Degrader ARV-471 | Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/arvinas-and-pfizer-announce-global-collaboration-develop (accessed on 2 November 2022).
- Accutar Biotech. Available online: https://www.accutarbio.com/workflow/ (accessed on 2 November 2022).
- Understanding Protein Degradation–Bristol Myers Squibb. Available online: https://www.bms.com/media/media-library/scientific-media-resources/understanding-protein-degradation-and-resources.html (accessed on 2 November 2022).
- Dialectic. Available online: https://www.dtsciences.com/ (accessed on 2 November 2022).
- Foghorntx Therapeutics Pipeline. Available online: https://foghorntx.com/pipeline/ (accessed on 2 November 2022).
- Therapeutic Pipeline. Available online: https://www.kymeratx.com/pipeline/ (accessed on 2 November 2022).
- Nurix Pipeline. Available online: https://www.nurixtx.com/pipeline/ (accessed on 2 November 2022).
- Targeted Protein Degradation–C4 Therapeutics. Available online: https://c4therapeutics.com/our-science/targeted-protein-degradation (accessed on 2 November 2022).
- Cullgen. Available online: https://www.cullgen.com (accessed on 3 November 2022).
- Mullard, A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250.
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697.
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C.; et al. Extended Pharmacodynamic Responses Observed upon PROTAC-Mediated Degradation of RIPK2. Commun. Biol. 2020, 3, 1–13.
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751.
- Wu, Y.; Yang, Y.; Wang, W.; Sun, D.; Zheng, M.; Zhou, Y.; Liang, J.; Zhu, M.; Li, H.; Chen, L. PROTAC Technology as a Novel Tool to Identify the Target of Lathyrane Diterpenoids. Biol. Med. Chem. 2022.
- Li, X.; Song, Y. Proteolysis-Targeting Chimera (PROTAC) for Targeted Protein Degradation and Cancer Therapy. J. Hematol. Oncol. 2020, 13, 50.
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as Emerging Anticancer Therapeutics. Oncogene 2020, 39, 4909–4924.
- Kargbo, R.B. PROTAC Molecules for the Treatment of Autoimmune Disorders. ACS Med. Chem. Lett. 2019, 10, 276–277.
- Kumar, D.; Hassan, M.I. Targeted Protein Degraders March towards the Clinic for Neurodegenerative Diseases. Ageing Res. Rev. 2022, 78, 101616.
- Powell, M.; Blaskovich, M.A.T.; Hansford, K.A. Targeted Protein Degradation: The New Frontier of Antimicrobial Discovery? ACS Infect. Dis. 2021, 7, 2050–2067.
- Desantis, J.; Goracci, L. Proteolysis Targeting Chimeras in Antiviral Research. Future Med. Chem. 2022, 14, 459–462.
- Trial of ARV-110 in Patients with Metastatic Castration Resistant Prostate Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03888612 (accessed on 8 November 2022).
- A Phase 1/2 Trial of ARV-471 Alone and in Combination with Palbociclib (IBRANCE®) in Patients with ER+/HER2- Locally Advanced or Metastatic Breast Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04072952 (accessed on 8 November 2022).
- Accutar Biotechnology Inc. A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Preliminary Anti-Tumor Activity of AC682 in Patients with Estrogen Receptor Positive/Human Epidermal Growth Factor Receptor 2 Negative (ER+/HER2-) Locally Advanced or Metastatic Breast Cancer. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- A Study of ARV-766 Given by Mouth in Men with Metastatic Castration-Resistant Prostate Cancer Who Have Progressed on Prior Approved Systemic Therapies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05067140 (accessed on 8 November 2022).
- Study to Evaluate the Safety and Tolerability of CC-94676 in Participants with Metastatic Castration-Resistant Prostate Cancer-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04428788 (accessed on 8 November 2022).
- A Study of DT2216 in Relapsed/Refractory Malignancies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04886622 (accessed on 8 November 2022).
- FHD-609 in Subjects with Advanced Synovial Sarcoma or Advanced SMARCB1-Loss Tumors-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04965753 (accessed on 9 November 2022).
- A Single and Multiple Ascending Dose Trial of KT-474 in Healthy Adult Volunteers and Patients with Atopic Dermatitis (AD) or Hidradenitis Suppurativa (HS)-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04772885 (accessed on 9 November 2022).
- Kymera Therapeutics, Inc. A Phase 1, Multicenter, Open-Label, Dose Escalation and Expansion Study to Evaluate the Safety, Tolerability, PK/PD, and Clinical Activity of Intravenously Administered KT-413 in Adult Patients with Relapsed or Refractory B-Cell NHL. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- Nurix Therapeutics, Inc. A Phase 1, Dose Escalation, Safety and Tolerability Study of NX-2127, a Bruton’s Tyrosine Kinase (BTK) Degrader, in Adults with Relapsed/Refractory B-Cell Malignancies. 2022. Available online: clinicaltrials.gov (accessed on 2 November 2022).
- A Study of NX-5948 in Adults with Relapsed/Refractory B-Cell Malignancies-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05131022 (accessed on 9 November 2022).
- A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05355753 (accessed on 9 November 2022).
- Effects of Systematic Proprioceptive-Tactile Stimulation with Use of the Protac MyFit®-Tabular View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04173871 (accessed on 9 November 2022).
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283.
- Gopal, P.; Dick, T. Targeted Protein Degradation in Antibacterial Drug Discovery? Prog. Biophys. Mol. Biol. 2020, 152, 10–14.
- Montrose, K.; Krissansen, G.W. Design of a PROTAC That Antagonizes and Destroys the Cancer-Forming X-Protein of the Hepatitis B Virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740.
- Lin, C. HCV NS3-4A Serine Protease. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.-L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; ISBN 978-1-904933-20-5.
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small Molecule Degraders of the Hepatitis C Virus Protease Reduce Susceptibility to Resistance Mutations. Nat. Commun. 2019, 10, 3468.
- Zhao, J.; Wang, J.; Pang, X.; Liu, Z.; Li, Q.; Yi, D.; Zhang, Y.; Fang, X.; Zhang, T.; Zhou, R.; et al. An Anti-Influenza A Virus Microbial Metabolite Acts by Degrading Viral Endonuclease PA. Nat. Commun. 2022, 13, 2079.
- Li, H.; Wang, S.; Ma, W.; Cheng, B.; Yi, Y.; Ma, X.; Xiao, S.; Zhang, L.; Zhou, D. Discovery of Pentacyclic Triterpenoid PROTACs as a Class of Effective Hemagglutinin Protein Degraders. J. Med. Chem. 2022, 65, 7154–7169.
- Xu, Z.; Liu, X.; Ma, X.; Zou, W.; Chen, Q.; Chen, F.; Deng, X.; Liang, J.; Dong, C.; Lan, K.; et al. Discovery of Oseltamivir-Based Novel PROTACs as Degraders Targeting Neuraminidase to Combat H1N1 Influenza Virus. Cell Insight 2022, 1, 100030.
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-COV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RiboTAC) Degraders. ACS Cent. Sci. 2020, 6, 1713–1721.
- Su, X.; Ma, W.; Feng, D.; Cheng, B.; Wang, Q.; Guo, Z.; Zhou, D.; Tang, X. Efficient Inhibition of SARS-CoV-2 Using Chimeric Antisense Oligonucleotides through RNase L Activation**. Angew. Chem. -Int. Ed. 2021, 60, 21662–21667.
More