Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mounira El Euch and Version 2 by Peter Tang.
Since the emergence of the COVID-19 pandemic at the end of 2019, a massive vaccination campaign has been undertaken rapidly and worldwide. Like other vaccines, the COVID-19 vaccine is not devoid of side effects. Typically, the adverse side effects of vaccination include transient headache, fever, and myalgia. Endocrine organs are also affected by adverse effects. The major SARS-CoV-2 vaccine-associated endocrinopathies reported since the beginning of the vaccination campaign are thyroid and pancreas disorders. SARS-CoV-2 vaccine-induced pituitary diseases have become more frequently described in the literature.
- pituitary
- SARS-CoV-2
- COVID-19
- vaccine
- hypophysitis
- apoplexy
- ASIA syndrome
- vaccine-induced thrombotic thrombocytopenia
- VITT
1. Introduction
Coronavirus disease 2019 (COVID-19) has been initially labeled as a severe potentially lethal respiratory infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. However, it has slowly become clear that many extra-pulmonary manifestations greatly contribute to the severity of the infection [2][3][2,3]. Furthermore, COVID-19 seems to have a wide-ranging impact on the body with little-known clinical features [4]. Data relating to the virus’ impact on the endocrine system is gaining momentum by the day. It has become an increasingly studied subject that sheds light on a variety of disorders, such as pituitary lesions, thyroid dysfunction, diabetes, adrenal insufficiency, and hypogonadism [5][6][7][5,6,7]. There is evidence that organs expressing the angiotensin-converting enzyme 2 (ACE2) receptor (such as the pituitary gland, thyroid, pancreas, adrenals, and gonads) can be the targets of the virus, as this receptor enhances the SARS-CO-2 attachment, permitting the infliction of cell damage [8][9][8,9]. COVID-19 infection can disrupt the functioning of endocrine organs and, similarly, COVID-19 vaccines can also induce endocrine dysfunction [5][10][11][5,10,11]. Multiple vaccines with variable efficacy and safety have been developed against COVID-19. Both the effect of the vaccine and the clinical course of the infection are inherently related to the systemic physiological response of the body. They remain also conditioned by the underlying health status of the patients [12]. The large use of vaccines worldwide (Pfizer–BioNTech, Oxford–AstraZeneca, Sinopharm BIBP, Moderna, Janssen, CoronaVac, Covaxin, Novavax, Convidecia, and Sanofi–GSK), allows for a broad evaluation of their side effects [13]. To date, the documented endocrine dysfunctions following COVID-19 vaccination concern primarily the thyroid gland, the pancreas’ beta cells, the adrenal glands, and the pituitary glands [5][7][5,7].
2. Hypophysitis
2.1. Pathogenesis
Hypophysitis is a heterogeneous condition attributable to an inflammation of the pituitary gland and its stalk. Inflammation can affect only the anterior pituitary, the posterior pituitary, or the entire gland resulting in panhypophysitis [14][43]. Hypophysitis can be broadly classified into primary and secondary forms. The precise etiology of primary hypophysitis remains unknown. It amounts to autoimmune and other inflammatory or infiltrative forms of isolated pituitary involvement [15][44]. Secondary hypophysitis occurs as a result of a reaction of the pituitary gland to local or systemic events, among which are local processes, systemic diseases, infections, neoplastic processes, and drugs [16][45]. Apart from etiological classification, hypophysitis can be presented histologically with various aspects. It has lymphocytic, granulomatous, xanthomatous, IgG-4 related, necrotizing, and mixed forms [14][43]. However, accurate classification is not always possible due to the overlap in pathological features. The presentation of hypophysitis induced by systemic inflammatory diseases does not differ from those induced by anti-COVID-19 vaccines. However, the pathophysiological mechanisms evoked are different. Several of these mechanisms cannot be proven directly since it is necessary to carry out a pathological analysis of the pituitary gland to allow this confirmation [17][46]. Since the pituitary is a gland that is very difficult to access for biopsy, performing such acts can lead to complications, particularly an aggravation of pre-existing deficits. This failure rate of biopsy is even higher in the event of isolated damage to the pituitary stalk [18][47]. Among the pathophysiological mechanisms of post-vaccine hypophysitis, autoimmune and inflammatory syndromes induced by vaccine adjuvants (ASIA) were the most cited [19][48]. Adjuvants are substances used to enhance the magnitude and durability of the immune response, which makes them an undeniable asset in the upgrading processes of vaccines. In genetically susceptible subjects, exposure to adjuvants may, on rare occasions, set off polygenic auto-immune diseases [20][49]. The immune disruption in such cases is attributed mainly to molecular mimicry, which triggers polyclonal activation of B lymphocytes [21][50]. The reviewed data on adjuvants used in COVID-19 vaccines showed that components such as aluminum salts, emulsions, oils, toll-like receptors, AS01B, four lipids of the mRNA vaccine, and polyethylene glycol might generate an immune response in susceptible individuals (Figure 1) [20][22][23][38,40,49].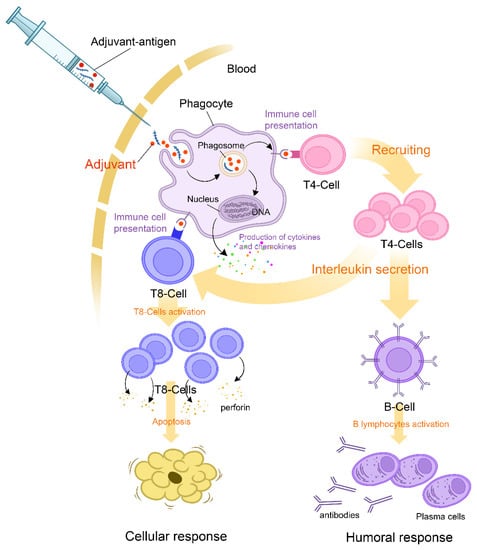
Figure 1.
Mechanism of action of adjuvants and initial triggers explaining the pathophysiology of the ASIA syndrome following COVID-19 vaccination.
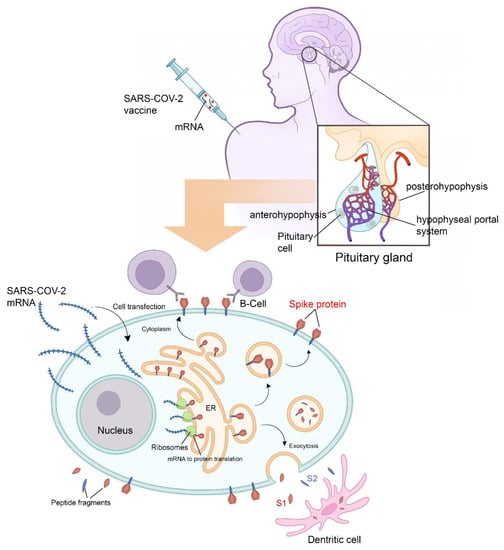
Figure 2. Physiology of pituitary cell protein’s expression and vaccine-induced hypophysitis pathophysiology. Following adjuvant internalization and mRNA release, the viral signal peptide drives antigen production in the endoplasmic reticulum (ER). After sorting in the Golgi network, S protein acquires its final position in the human cell membrane, where S1 is exposed to the extracellular space. Antigen sorting and trafficking may also induce the release of S protein-containing exosomes. Also shown are dendritic cells (professional antigen-presenting cells) engulfing circulating antigens, and antibody-mediated binding of B cells to cell-anchored antigens. All the above mentioned mechanisms potentiate the inflammatory mechanisms. All the Endocrine consequences after hypophysitis are mainly hypogonadism, hypothyroidism, hypoadrenalism and diabetes insipidus.
2.2. Clinical Presentations
Hypophysitis typically presents with symptoms that can be attributed either to pituitary deficiencies or to the mass effect of an enlarged pituitary gland and infundibulum. Symptoms related to the mass effect are quite common: visual disturbances concern 10% to 30% of patients with primary hypophysitis, while headaches affect approximately half of them [14][43]. The etiology, severity, and extent of the pituitary damage are the main factors dictating the loss of anterior and/or posterior pituitary hormones and, therefore, conditioning the clinical presentation. For instance, when anterior pituitary dysfunction is associated with DI, the damage is most likely due to an inflammatory process or to metastasis, as this clinical presentation is highly improbable in pituitary adenomas [15][44]. In vaccine-induced hypophysitis, inflammatory processes predominantly and equally affect corticotrophs, gonadotrophs, and thyreotrophs. Most importantly, this pattern is similar to other classical hypophysitis, unlike in pituitary adenomas which rarely affect the corticotroph axis [24][58]. All patients require a complete pituitary function testing, including the following: an 8 am cortisol and/or adrenocorticotropin stimulation test; TSH; free thyroxine; prolactin; IGF-1; LH; and FSH with estradiol in premenopausal women or testosterone in men. If DI is suspected, the levels of serum sodium should be determined along with plasma and urine osmolarity. Hyponatremia caused by adrenal insufficiency or severe hypothyroidism should be ruled out. Depending on the extension of the inflammation and the clinical context, each case may have systemic features related to the severity of tissue damage [16][45]. In the studied cases, clinical presentations varied from a single axis deficiency in one patient (isolated corticotroph deficiency) to multiple pituitary deficits. Numerous neuro-radiological findings have been reported in vaccine-induced hypophysitis [25][59]. The main features suggestive of the disease include a diffuse and symmetric mild-to-moderate pituitary gland enlargement and/or a thickened, non-deviated stalk. Contrast uptake is usually intense and generally homogeneous [26][60]. In infundibulo-neurohypophysitis, stalk thickening is isolated (without features of gland enlargement); the loss of posterior pituitary bright spot can also be seen and is attributed to the depletion of vasopressin granules [27][24]. Images of an enlarged pituitary gland may mimic a pituitary adenoma, leading to misdiagnosis [26][60]. Contrariwise, in advanced stages of the disease, an atrophic aspect of the pituitary gland can be observed along with images of sellar arachnoidocele or empty sella [28][61]. Despite lacking the specificity for diagnosing vaccine-induced hypophysitis, MRI findings can help establish a diagnosis in the right clinical context with a major benefit of being a noninvasive method. For example, proof of stalk thickening in a woman who has recently been vaccinated and is presenting with headache and hypopituitarism is highly suggestive of hypophysitis [29][34].2.3. Treatment
Owing to their anti-inflammatory properties, glucocorticoids (GCs) are regarded as the treatment cornerstone of hypophysitis [15][44]. In the chronic phases, however, when irreversible damages take place, anti-inflammatory treatment may not affect radiological or hormonal outcomes. Moreover, spontaneous resolution of pituitary infiltration with or without permanent pituitary dysfunction did occur in some cases [14][43]. In vaccine-induced hypophysitis, the evolution remains unclear. Occasionally, the process persisted up to several months, requiring a regular follow-up [29][34]. In a German cohort, radiological improvement was noted in 46% of patients with primary hypophysitis who were managed through observation only. Additionally, one third of the patients had hormonal recovery, mainly including vasopressin and ACTH [30][62]. It is still uncertain whether GCs are superior to simple observation in allowing for a better pituitary function recovery or not, as no randomized controlled studies have been performed yet. Given the broad spectrum of GC side effects, a consideration of the risks and benefits is crucial when deciding whether to actively treat mild cases of primary hypophysitis or not. Clinical signs and symptoms should be taken into consideration when selecting a treatment option. For example, in patients with mild-to-moderate headache, mild pituitary dysfunction, and no mass effect on optic chiasm, observation may be safely envisaged [14][15][43,44]. Hormonal replacement of the other pituitary axes is also necessary. Hormonal replacement doses in vaccine-induced hypophysitis do not differ from other pituitary diseases in the reported cases. Essentially, for DI, vasopressin replacement should be maintained in case of persistent polyuro polydipsia. Patient education remains important in order to avoid a decompensation of these hormonal deficiencies. Tumoral compression on neighboring structures is rarer in hypophysitis than in the case of pituitary apoplexy [31][63]. In such cases, glucocorticoids will seemingly decrease the inflammatory process. In addition, a surgical intervention could be considered in order to outperform a debulking to save the optic chiasma. The followed patients did not mention any mechanical compression that could compromise the visual prognosis or the integrity of cranial nerves.3. Pituitary Apoplexy
3.1. Pathogenesis
The Oxford–AstraZeneca COVID-19 vaccine is a viral vector vaccine using the modified adenovirus ChAdOx1. The vaccine contains a full-length coding sequence of SARS-CoV-2 spike protein in the form of DNA. When the viral vectors infect the host cells, the aforementioned genetic code is transcribed, translated, and then presented as a spike protein to the body’s immune system by the host cells. The ChAdOx1 nCov-19 vaccination produced a positive immune reaction in all of the patients with apoplexy [32][64]. Following several large vaccine campaigns, there was mounting evidence of serious adverse effects related to this vaccine, including thrombosis and bleeding. This syndrome has been termed “Vaccine-Induced Thrombotic Thrombocytopenia” (VITT) [33][65]. It occurred more frequently in young women and was imputed only to viral vector vaccines. VITT seems to be a phenomenon similar to heparin-induced thrombocytopenia and appears to have an autoimmune provenance [34][66]. Currently, five criteria are used to define VITT, including recent vaccination, thrombosis, thrombocytopenia, elevated D-dimer levels, and positivity of anti-PF4 antibodies (Figure 3) [35][67].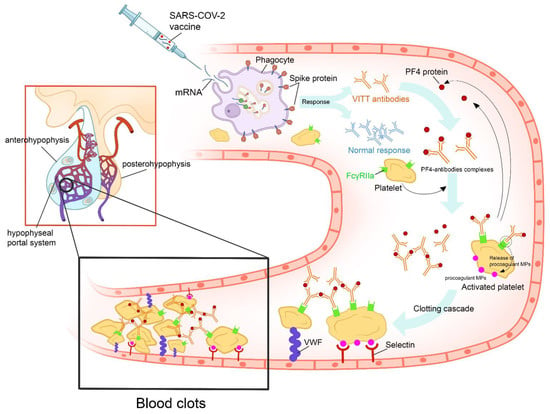
Figure 3. Proposed figure showing the cascade of pathogenic events that could favor the onset of vaccine-induced immune thrombotic thrombocytopenia in individuals vaccinated with anti-COVID-19 vaccines. After the administration of the vaccine, the recipient’s cells produce harmless COVID-19 proteins and the immune system responds by producing protective antibodies. In some cases, the vaccine’s adjuvants and spike proteins trigger a type I interferon response and the production of VITT antibodies. VITT is caused by antibodies that recognize PF4 bound to platelets. These antibodies are IgGs that activate platelets via low-affinity platelet FcγRIIa receptors (receptors on the platelet surface that bind the Fc portion of IgG). Anti-PF4 antibodies cause cellular activation that, besides activating platelets and coagulation reactions, activates monocytes, neutrophils, and endothelial cells (leading to tissue factor expression). Activation of these other cell types further contributes to the high thrombosis risk accelerated by other factors, such as VWF and procoagulant MP. All of these mechanisms will lead to pituitary apoplexy. (PF4: platelet factor 4; IgG: immunoglobulin G; VITT: vaccine-induced immune thrombotic thrombocytopenia; VWF: Van Willebrand Factor; MP: microparticles).