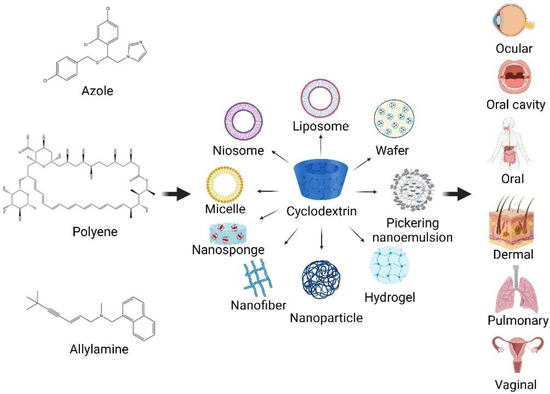
1. Cyclodextrins (CDs) -Based Nanocarriers in Enhanced Antifungal Delivery
Presently, nanotechnology offers an ingenious solution for overcoming the drawbacks of poorly water-soluble drugs in formulation development. The use of several CD-based nanocarriers in formulations has been reported for drug delivery systems (Figure 13). The combined technology has also been of interest as an effective system for antifungal drug delivery. The use of biocompatible and biodegradable components, as well as versatility, stability, high drug loading, and permeability through biological membranes, are obvious benefits of using these carrier systems for the effective delivery of antifungal agents.
Figure 13.
CD-based nanocarriers for antifungal agents in drug delivery systems (created with Biorender.com).
The following are case studies of CD-based formulations for antifungal drug delivery. The effects of CDs on enhancing the solubility and permeability of drugs through various routes of administration, leading to enhanced bioavailability and therapeutic antifungal activity, are also discussed.
2. Oral Drug Delivery
Oral dosage forms are recognized as the most acceptable formulations for patients. However, due to low aqueous solubility, poor permeability, chemical instability, degradation in the gastrointestinal tract, extensive metabolism, and the unpleasant organoleptic properties of drugs, the oral route is still challenging for developing pharmaceuticals. To overcome these drawbacks, CDs were used to increase the solubility and enhance the dissolution rate of poorly water-soluble drugs
[1][79].
Kumar et al. (2014)
[2][80] studied the effect of CD on the solubility enhancement of itraconazole. Firstly, itraconazole sulfate salt was synthesized and prepared in physical mixtures with βCD and HPβCD. The physical mixtures resulted in an enhanced dissolution rate in simulated gastric fluid (pH 1.2) compared with itraconazole salt and pure itraconazole. Of these, the dissolution rate of the mixture with HPβCD was higher than that of βCD. The authors suggested that the developed itraconazole sulfate/CD mixtures could be potentially applied to oral drug delivery systems.
Floating drug delivery systems are one of the most widely used approaches to increase the gastric retention time of oral dosage forms. Among the single and multiple-unit systems, multiple-unit particulate dosage forms containing microspheres show more benefits due to their uniformity when passing along the gastrointestinal tract, preventing gastric emptying and providing controlled release, thereby decreasing the variability in drug absorption and reducing local irritation
[3][81]. Itraconazole, an orally administered antifungal compound, was loaded in microspheres using an ionotropic gelation technique with chitosan (1.5–2.5%
w/
v). The resulting microspheres containing 1%
w/
v itraconazole were lyophilized and further developed into floating tablets. The in vitro permeation was studied at different pH values, i.e., pH 5, 6, and 7.4. The results revealed that the highest permeability was observed from a formulation composed of 4.72%
w/
v itraconazole/RMβCD, 2%
w/
v chitosan, 3%
w/
v polyethylene glycol, and 1%
w/
v tripolyphosphate at a pH of 5. The optimized formulation was evaluated for in vivo buoyancy activity with gamma scintigraphy using rabbits and showed it remained floating in the stomach for 6.5 h and had improved bioavailability
[4][20].
Due to its poor aqueous solubility, posaconazole has been administered as an oral suspension. To enhance the solubility of posaconazole, a posaconazole/HPβCD inclusion complex was prepared and characterized by Tang et al. (2016)
[5][47]. The solubility of posaconazole was greatly enhanced (i.e., 82 times), and a high dissolution (>90%) was obtained when complexed with HPβCD at different pH levels representing gastrointestinal tract conditions. The in vitro antifungal susceptibility study also proved that complex was the most susceptible to
Candida albicans, and nearly 97% of fungi were inhibited by ≤1 µg/mL under the minimal inhibitory concentration (MIC) at which 90% of the isolates were inhibited (MIC90) with 0.06 µg/mL. In the case of the least susceptible
Aspergillus niger, 97% of the MICs were ≤4 µg/mL, and the MIC90 was at 1 µg/mL. Their findings revealed that HPβCD was able to enhance the aqueous solubility and dissolution rate in simulated gastric conditions, thereby maintaining a high susceptibility to the tested fungal species.
3. Oral Local Drug Delivery
In the oral cavity, candidiasis is the most common fungal infection. Its clinical manifestations range from relatively minor disorders, such as acute pseudomembranous forms (e.g., oral thrush), to chronic erythematous and hyperplastic forms, as well as systemic superinfections among immunocompromised patients, with mortality rates of 30 to 50%
[6][82]. Despite the availability of various effective antimycotics to treat oral candidiasis, therapeutic failure is common due to the oral cavity’s peculiar features, tending to lower local drug concentrations to subtherapeutic levels
[7][83].
Solid sponge-like matrices (wafers) obtained by lyophilizing polymer solutions offer advantages over conventional drug delivery systems; for example, they can maintain their swelling gel structure for extended periods of time within the oral cavity
[8][84]. In the study of Mura et al. (2015)
[9][85], they prepared an econazole/SBEβCD/citric acid ternary complex to improve the solubility of econazole. The binary, as well as the ternary complexes, provided a significantly higher antimycotic efficacy against selected
Candida strains compared with that of econazole alone. Again, the ternary complex, providing high solubility as well as dissolution efficiency, was selected and loaded into low methoxy amidated pectin and carboxymethylcellulose to develop a lyophilized wafer formulation. The therapeutic antifungal efficacy using a time-killed assay was also tested in vitro for
Candida albicans and
Candida krusei, and resulted in 30.99 ± 1.23 h and 33.01 ± 4.01 h, respectively. The developed wafer formulation did not affect or lessen the antifungal activity of econazole when compared with that of the ternary system.
Antifungals are also delivered by their incorporatiation into medicated chewing gum, which is a convenient delivery system for fungal infections in the oral cavity
[10][86]. Econazole and miconazole solubilities were enhanced by complexing with CDs
[11][87]. Then, both the drug/CD complex, as well as free drugs, were incorporated into the chewing gum, which was prepared by mixing a Fertin gum base, solid paraffin wax, and other excipients. In the case of econazole, the econazole/βCD complex only moderately increased the release of econazole compared with the release from the neat econazole gum. The miconazole/HPβCD complex-loaded gum had a much higher drug release (25% within 30 min) than the miconazole gum. When compared with neat miconazole chewing gum, miconazole/HPβCD chewing gum demonstrated superior antifungal activity due to the increased drug release.
For decades, advanced research on drug delivery systems has focused on enhancing therapeutic effects. Nanofibers have also gained attention in drug delivery because of their numerous advantages, including their high loading and encapsulation of drugs and great flexibility for incorporating a wide range of materials
[12][13][88,89]. Fast-dissolving nanofiber mats were optimized using a polyvinyl pyrrolidone (PVP)/HPβCD nanofiber for clotrimazole delivery in the oral mucosa
[14][90]. CD could increase drug dissolution in the saliva, thereby facilitating drug absorption and resulting in enhanced drug bioavailability. For treating oral candidiasis, both rapid antifungal activity and prolonged contact time to reach the MIC are required. Because the resultant PVP/HPβCD nanofibers displayed fast drug release, modified coated nanofibers with chitosan and polyvinyl alcohol were employed to increase the contact time with the oral mucosa. The modified clotrimazole nanofibers demonstrated good mucoadhesive and controlled-release properties and antifungal activity against
Candida albicans [15][91].
4. Vaginal Drug Delivery
In recent years, vaginal infections have become the most common gynecologic disorder in which various pathogens, such as fungi, viruses, and protozoa, cause various diseases in the female reproductive tract. Among the pathogenic fungi,
Candida albicans is still considered the most prevalent fungal pathogen responsible for vulvovaginal candidiasis. Because of the gastric pH and the extensive first-pass metabolism for drugs taken orally, as well as to avoid systematic side effects, the vaginal route has been preferred to administer antifungals for local therapy
[16][92]. However, the dynamic elimination mechanisms in the vaginal lumen cause insufficient residence time at the target absorption site, causing an uneven distribution of drugs onto the vaginal mucosa
[17][93]. Traditional vaginal drug delivery systems are unable to maintain an effective drug concentration for an extended period of time. In fact, antifungals for vaginal medications are required to be solubilized and retained at or near the mucosa for a long residence time and be able to penetrate the vaginal tissue to improve their therapeutic efficacy. Numerous investigations have reported that CDs act as solubilizers and permeation enhancers when incorporated into tablets, mucoadhesive gels, creams, and films for vaginal antifungal delivery.
Natamycin is a poorly water-soluble compound. Solubility enhancement was achieved by complexation with γCD with a stability constant of 667 M
−1 (Table 2). This complex provided a similar MIC when compared with the pure drug against
Candida. Furthermore, the natamycin/γCD complex-loaded bioadhesive tablet, based on Carbopol 934P, provided good mucoadhesion as well as prolonged drug release
[18][53]. Another case study of the itraconazole bioadhesive vaginal tablet was developed by Cevher et al. (2014)
[19][94]. The aqueous solubility of itraconazole was greatly enhanced with SBEβCD, and the resultant complex exhibited the highest efficacy, i.e., the lowest MIC against
Candida albicans among the tested complexes. Additionally, the itraconazole/SBEβCD complex incorporated in the Carbopol 934P-based bioadhesive tablet provided good mucoadhesion and prolonged release of itraconazole for 36 h.
Another polyene antibiotic, amphotericin B, is also therapeutically effective in vaginal candidiasis. However, its application is limited due to its poor aqueous solubility and toxicity. In the study of Kim et al. (2010)
[20][95], they improved the aqueous solubility of amphotericin B using HPβCD or HPγCD. The amphotericin B/HPγCD complex, which demonstrated a significantly high solubility of amphotericin B, was further loaded in an in situ gel based on thermosensitive multiblock copolymers. These copolymers were synthesized from Pluronic
® triblock copolymers (Pluronic
® P85 and P104) and di-(ethylene glycol) divinyl ether. The developed formulation underwent a sol-gel transition at the site of action. Moreover, the developed amphotericin B/HPγCD complex-loaded in situ gel showed controlled-release characteristics and was less toxic than free amphotericin B in the HEK 239 cell line. In a histopathological study in mice, no visible signs of inflammation or necrosis in the vaginal mucosa were observed.
Deshkar et al. (2019)
[21] developed a voriconazole/HPβCD complex-loaded in situ gel for vaginal delivery. Due to the solubility of voriconazole being enhanced in the presence of HPβCD, a significantly higher drug release was observed compared with the voriconazole gel without HPβCD. A further in vivo vaginal tissue uptake study was conducted in Wistar rats. It was demonstrated that the HPβCD-based in situ gel formulation provided a higher drug uptake in the vaginal tissue than an in situ gel without the HPβCD and voriconazole dispersion.
5. Pulmonary Drug Delivery
Pulmonary fungal infections are a major health problem, particularly in immunocompromised patients with lung cancer, cystic fibrosis, human immunodeficiency virus (HIV), etc.
[22][96]. Conventional treatments in pulmonary delivery are limited due to several factors, including bypassing the upper airways to reach the lungs, the branching structure of the lungs resulting in difficult access to the alveolar regions, and the rapid blood turnover in the lungs. Another factor is the presence of a thick mucus gel layer in which the inhaled drugs could be entrapped and then removed by mucociliary clearance. As a result, a large dose is usually required for a long period of time to cause toxicity
[22][23][96,97].
CDs were shown to improve the bioavailability of poorly water-soluble drugs in aerosols or solutions for pulmonary delivery
[24][98]. Yang et al. (2010)
[25][99] prepared an itraconazole/HPβCD complex solution for pulmonary delivery using nebulization. An itraconazole nanoparticle colloidal dispersion composed of mannitol and lecithin was developed using the ultra-rapid freezing method for comparison. Both the itraconazole/HPβCD solution and colloidal dispersion were suitable for deep lung drug delivery in mice. The itraconazole/HPβCD complex solution demonstrated a faster systemic absorption of itraconazole across the lung epithelium than that of a colloidal dispersion. This is possible because itraconazole is released from the nanoparticles and is required for the phase-to-phase transition. However, from the pharmacokinetic data, the nanoparticulate itraconazole colloidal dispersion exhibited an enhanced drug bioavailability because of the presence of lecithin as an additional permeation enhancer. The authors suggested that the uncertain stability and the regulatory status of HPβCD should be considered in pulmonary administration.
Another research group approached treating invasive fungal infections by preparing a voriconazole/SBEβCD complex for targeted drug delivery to the lungs using nebulization
[26][100]. The high solubilization capacity of voriconazole by SBEβCD in the developed aerosol solution resulted in high drug loading, leading to enhanced voriconazole concentrations in the lung tissues and plasma after single and multiple doses of inhaled administration. Hence, the pharmacokinetic data of inhaled aqueous solutions of voriconazole confirmed that effective therapeutic outcomes could be achieved using a voriconazole/SBEβCD solution.
6. Ocular Drug Delivery
Ocular drug delivery is a considerable challenge in developing pharmaceutical formulations because of the complex anatomy of the eyes. The anatomical and physiological barriers in the eyes, such as tear turn over, nasolacrimal drainage and blinking of the eyes, etc., limit the bioavailability of drugs in ocular tissues. To obtain the optimum penetration, drugs need to be solubilized in lachrymal fluid and pass the tear film barrier
[27][101]. The use of solubilizers, as well as permeation enhancers such as CDs could help to achieve therapeutic drug concentrations in various parts of ocular tissues.
Mahmoud et al. (2011)
[28][102] developed econazole/SBEβCD complex-loaded chitosan nanoparticles using the ionic gelation method. Ionically crosslinked chitosan/SBEβCD nanoparticles acted as an interesting chitosan-based delivery system for econazole to the ocular membrane. The developed formulations showed particle sizes in the nanometer range (90 to 673 nm) and positive zeta potential values (+22 to +33 mV). The optimum formulation provided good mucoadhesive properties in a controlled-release manner. The antifungal activity of the econazole-loaded chitosan/SBEβCD nanoparticles showed better efficacy when compared with an econazole solution. Regarding these observations, the authors concluded that the optimized formulation might exhibit a potential carrier for ocular drug delivery.
A voriconazole/HPβCD complex-loaded hydrogel was developed by Diaz-Tome et al. (2021) to treat fungal keratitis
[29][43]. The solubility of voriconazole was greatly increased using HPβCD. Furthermore, they prepared two types of gels, i.e., an ion-sensitive hydrogel prepared with a voriconazole/HPβCD-loaded kappa-carrageenan hydrogel and a Vfend
®-loaded (a commercial intravenous voriconazole/SBEβCD preparation) hyaluronic acid hydrogel. The ex vivo permeability study showed that both hydrogels provided good corneal permeability. These formulations increased the residence time on the ocular surface and proved to be nonirritants. These formulations could serve as suitable alternatives to treat fungal keratitis.
Chaudhari et al. (2022)
[30][40] formulated a ketoconazole/SBEβCD inclusion complex-loaded in situ gel for ocular fungal infections. The aqueous solubility of ketoconazole was enhanced by forming an inclusion complex with SBEβCD. The ketoconazole/SBEβCD complex was further loaded in a thermosensitive in situ gel. The drug release was increased 10-fold compared with the intact ketoconazole. The in situ gel exhibited a more sustained drug release because of the diffusion of drugs from the polymer matrix. The ex vivo permeation through a goat corneal membrane revealed that the permeation flux of ketoconazole from the formulation in the presence of SBEβCD (∼19.11 µg/cm
2/h) was significantly higher than that in the absence of SBEβCD (∼1.17 µg/cm
2/h). The formulations were nontoxic to human corneal endothelial cells and, thus, could potentially treat fungal keratitis. Another example is fluconazole/HPβCD complex-loaded Eudragit nanoparticles and chitosan-coated niosomal vesicles that are further incorporated into in situ gels
[31][70]. The developed formulation resulted in nanosized particles with high positive zeta potentials and high entrapment efficiencies. It also exhibited sustained release within 24 h, enhancing the corneal permeation without irritating the ocular surface and promising antifungal activity against
Candida albicans. The modified formulations using combined strategies, i.e., CD inclusion complexes, nanoparticulates, and in situ gels, provide the optimum eye drops for ocular delivery.
Pickering emulsions are surfactant-free emulsions stabilized by solid particles. Their unique structures endow them with good stability, excellent biocompatibility, and environmental friendliness. CD-based Pickering emulsions exhibited high safety and biocompatibility due to fewer quantities of stabilizers and were more stable against coalescence and separation when compared with conventional emulsions
[32][103]. Recently, amphotericin B-loaded CD-based Pickering nanoemulsions were developed by Maw et al. (2022)
[33][104]. This was achieved using αCD/medium chain triglyceride crystals as solid particle stabilizers at the oil and water interface. In addition, the solubility of amphotericin B was also increased by complexing with γCD and HPγCD. The developed formulations exhibited less amphotericin B aggregation and fewer hemolytic properties than commercial products (i.e., the amphotericin B-containing sodium deoxycholate micellar formulation). Moreover, sustained drug release was observed due to the high entrapment efficacy. The in vitro antifungal activity study also showed a better result in amphotericin B-loaded nanoemulsions than that of intact amphotericin B, and also comparable with that of commercial products against
Candida albicans. The developed CD-based Pickering nanoemulsions were physically and chemically stable over six months, which was greater than their respective non-Pickering nanoemulsions.
7. Dermal Drug Delivery
Topical antifungal drug delivery is one of the major routes to treat skin diseases associated with fungi. The route ensures direct access and a higher retention rate at the infected area on the skin
[34][105]. Furthermore, topical delivery also decreases systemic side effects and avoids first-pass metabolism. Che et al. (2015)
[35][106] developed a CD-based microemulsion loaded with ketoconazole for skin delivery. In the comparative study with the microemulsion without CD, the ketoconazole/HPβCD complex, ketoconazole aqueous suspensions, as well as the marketed product (ketoconazole cream, Daktarin Gold
®), the incorporation of HPβCD in the microemulsions provided a better retention time and enhanced permeation through the skin layer. The synergistic skin targeting effect might have been due to the high solubilization effect of HPβCD, which was able to promote drug deposition within the skin, as well as drug penetration through the skin. The in vitro antifungal activity study revealed that ketoconazole/HPβCD complex-loaded microemulsions resulted in the largest zone of inhibition against
Candida parapsilosis. Therefore, it could be concluded that a microemulsion combination with HPβCD may be a promising approach for the skin-targeted delivery of ketoconazole.
Deformable liposomes have also emerged to improve their effectiveness as drug carriers to increase drug penetration through biological mucous membranes due to their high flexibility and elasticity. These systems can squeeze themselves between the stratum corneum cells and reach deep skin layers. Itraconazole/HPβCD-loaded deformable liposomes were investigated by Alomrani et al. (2014)
[36][107]. HPβCD was incorporated to increase the solubility of drugs and to enhance skin penetration. The presence of CD provided physical stability to the developed liposome. In addition, higher deformability was observed, leading to good permeation towards the deep skin layer when compared with conventional liposomes. The in vitro antifungal activity revealed that the deformable liposome was active against
Candida albicans.
The potential of CD-based Pickering emulsions as a carrier of econazole for topical drug delivery was introduced by Leclercq and Nardello-Rataj (2016)
[37][108]. An econazole-loaded Pickering emulsion was developed using αCD, βCD, and γCD with paraffin oil and isopropyl myristate. Of these, αCD resulted in the most stable Pickering emulsion, followed by βCD and γCD due to the suitable affinity between the hydrophobic alkyl tail of the oil molecule and the small αCD cavity. The resulting Pickering emulsions were stable against the coalescent due to the formation of a dense film of oil/CD complex particles at the oil and water interface. The antifungal activity results displayed that the Pickering emulsions stabilized with αCD and βCD were able to inhibit
Candida albicans growth.
Recently, CD nanosponge, a novel nanotechnology platform containing CD was applied in the pharmaceutical field.. CD nanosponges are nanostructured materials with nanometer-sized cavities that are easily prepared using crosslinkers, such as carbonyl diimidazole or diphenyl carbonate
[38][39][109,110]. Srivastava et al. (2021)
[40][111] developed an econazole-loaded nanosponge comprising βCD and N,N-carbonyl diimidazole as a crosslinker. The optimized econazole nanosponge was further loaded in a topical hydrogel in which Carbopol 934P was used as a gelling agent. The developed nanogel containing the econazole-loaded nanosponge achieved a higher penetration of econazole across the skin barrier with a flux of 314 µg/cm
2 than the commercial product with a flux of 88 µg/cm
2 during the 24 h study. The nanogel showed better in vitro antifungal activity against
Candida albicans and in vivo antifungal activity in male albino Wistar rats than the commercially marketed product. These promising results might have been due to the function of the nanosponge, which helped econazole to permeate the skin layer and provided the controlled release of econazole for longer periods of time during the
res
earchtudy.
8. Parenteral Drug Delivery
Invasive fungal infections have been recognized as a major cause of morbidity and mortality in immunocompromised patients. The parenteral route is preferred for the delivery of compounds with a narrow therapeutic index and poor bioavailability and for the prescription of unconscious patients. To maintain therapeutic effectiveness, frequent injections are required, leading to patient discomfort. Therefore, for parenteral antifungal drug delivery, developing formulation technologies is considered important to provide a targeted therapeutic level and sustained release of drugs in a predictable manner
[41][112].
Posaconazole is a triazole antifungal agent with an extended spectrum of antifungal activity. However, its pharmacological effects are limited owing to its poor aqueous solubility and low bioavailability. The enhancement of the bioavailability of posaconazole was obtained with the complexation with SBEβCD, reported by Wang et al. (2018)
[42][113]. The solid posaconazole/SBEβCD complex, prepared by lyophilization, showed an excellent dissolution profile within 5 min. In addition, in vivo pharmacokinetic parameters were studied in rats after pure posaconazole was solubilized in cosolvents, and posaconazole/SBEβCD complexes were injected intramuscularly. The data showed that the posaconazole/SBEβCD complex significantly increased the peak concentration and bioavailability compared with pure posaconazole. The authors suggested that SBEβCD played an important role in stabilizing the saturated drug solution and preventing the precipitation of drugs at the injection site.
Mutlu-Agardan et al. (2020)
[43][114] studied an amphotericin B and amphotericin B/αCD complex double-loaded liposome to treat invasive fungal infections. Among the CDs tested, the highest solubilization was achieved with αCD, and this complex was further loaded into liposome preparations. The drug-in-CD-in-liposomes improved the physical stability and exhibited a higher percentage of entrapment efficiency, i.e., 80%, than only free amphotericin B-incorporated liposomes. Additionally, the novel amphotericin B double-loaded liposome formulation provided a rapid onset time followed by a sustained release property within 72 h. The antifungal activity against
Candida albicans showed that the amphotericin B double-loaded liposome exhibited significantly lower MIC and minimum fungicidal concentration (MFC) values when compared with the conventional and commercial products.
In general, CDs are used to enhance the aqueous solubility of antifungals and to overcome the limitations of nanoparticulate drug delivery systems, which include improving drug loading and entrapment efficiency. Another beneficial finding regarding CD-containing oral solid dosage forms is the enhancement of drug dissolution in aqueous media. Moreover, CD aids in decreasing the degradation of labile drugs and preserving antifungal potencies. Regarding toxicological issues, the drug-induced hemolysis of, for example, amphotericin B was significantly lowered with CD complexation. Additionally, it was noted that the incorporation of CD in formulations resulted in modified drug release, enhanced drug permeation through biological membranes, improved pharmacokinetics, and consequently increased the bioavailability of antifungals.