Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Zhaohui Luo.
Glioma is a life-threatening malignancy, and traditional radiotherapy and chemotherapy are not very effective. A growing number of studies have shown that microorganisms and their derivatives can be used as cancer therapies.
- microorganism
- targeted therapy
- gut–brain axis
- intestinal flora
1. Derivatives of Microorganisms Are Used to Treat Gliomas
Bacteria can produce a range of bioactive substances that are used in the treatment of gliomas through different mechanisms, such as bacteriocins, bacterial toxins, and enzymes.
Bacteria use ribosomes to synthesize bacteriocins and antimicrobial peptides that prevent other bacterial strains from invading their own ecological niches. Bacterial peptides can also prevent the growth of tumor cells. Prodigiosin causes cell death by activating the JNK pathway and reducing the AKT/mTOR pathway in glioblastoma (GBM) cells [18][1]. As a PAMP, flagellin is recognized by TLR and activates the body’s immune system. Flagellin can reduce the number of myeloid-derived suppressor cells (MDSCs) in tumor tissues and adjust the conversion of tumor-associated macrophages (TAMs) from M2 to M1 type. Therefore, it promotes the immune response in the glioma microenvironment and inhibits tumor growth [19][2]. Someone combined them and found that they significantly reduced the size of intracranial tumors in mice. P53 is a proapoptotic transcription factor. TSA leads to p53 phosphorylation by activating p38 mitogen-activated protein kinase (p38MAPK). TSA inhibits NF-κB activity by inducing IKK dephosphorylation [21][3]. TSA also inhibits GBM vascular proliferation [32][4]. MOX is able to induce cell apoptosis by increasing the Bcl-2-associated X protein/B-cell lymphoma 2 ratio and activating the caspase-3/-9 cascade in glioma, and it induces G0/G1 cell cycle arrest and apoptosis to inhibit the viability of glioma cells [22][5].
Bacteria express and release specific toxins, which are a class of highly poisonous proteins generated and released by bacteria with specific functions. Due to their great toxicity, bacterial toxins have been demonstrated to be effective cancer treatments. PE, the most aggressive virulence factor produced by Pseudomonas aeruginosa, can be employed to destroy tumor cells by inhibiting protein synthesis via ADP-ribosylation of eukaryotic elongation factor 2. PE can be combined with antibodies or receptor ligands for precise tumor targeting to generate chimeric proteins, and this toxin-linked targeting element forms an immunotoxin that can be exploited for cancer therapy [33][6]. The targeting element is responsible for attaching tumor surface molecules, while the toxin kills cancer cells. Vibrio cholerae produces cholera toxin, cholera toxin subunit B (CTB), which facilitates glioma-targeted drug delivery by targeting sphingolipid GM1 expressed in the blood–brain barrier (BBB), neovascularization, and glioma cells [24][7]. E. coli produces CNF1, which causes glioma cells to overexpress p21 and p16, promotes glioma cell senescence, blocks tumor progression and migration, and protects the structure and function of healthy surrounding tissues [25][8]. CTX, a peptide derived from scorpion venom, penetrates the BBB and preferentially recognizes and targets glioma cells; therefore, a group of researchers developed a chimeric protein of CTX-CNF1 that dramatically prolongs the survival time of glioma mice following systemic injection [26][9].
Essential amino acids are necessary for cell growth and cellular metabolism, and their depletion is one of the cancer treatments. Arginine deiminase (ADI) derived from Streptococcus pyogenes depletes arginine from tumor cells, resulting in a nutritional deficiency and inhibiting tumor cell development. The addition of suberoylanilide hydroxamic acid (SAHA) to ADI can further increase the strong tumorolytic effect [27][10]. Recent studies have demonstrated that the therapeutic effect of Arg deprivation is largely dependent on the expression of argininosuccinate synthase in tumors, regardless of whether they are of the nutrition-deficient type [34][11], but this therapy is not hindered by the BBB, provides multiple mechanisms for inducing apoptosis in glioma cells, and has enormous research potential.
To date, temozolomide is the only first-line chemotherapeutic agent for high-grade glioma, but drug resistance in tumors is also a significant problem, and the development of new drugs is a focus of current research. The derivatives of these microorganisms mentioned above have achieved a good effect against glioma in animal experiments and cellular experiments.
Thousands of microorganisms cause problems in humans while potentially contributing to drug development (Figure 1).
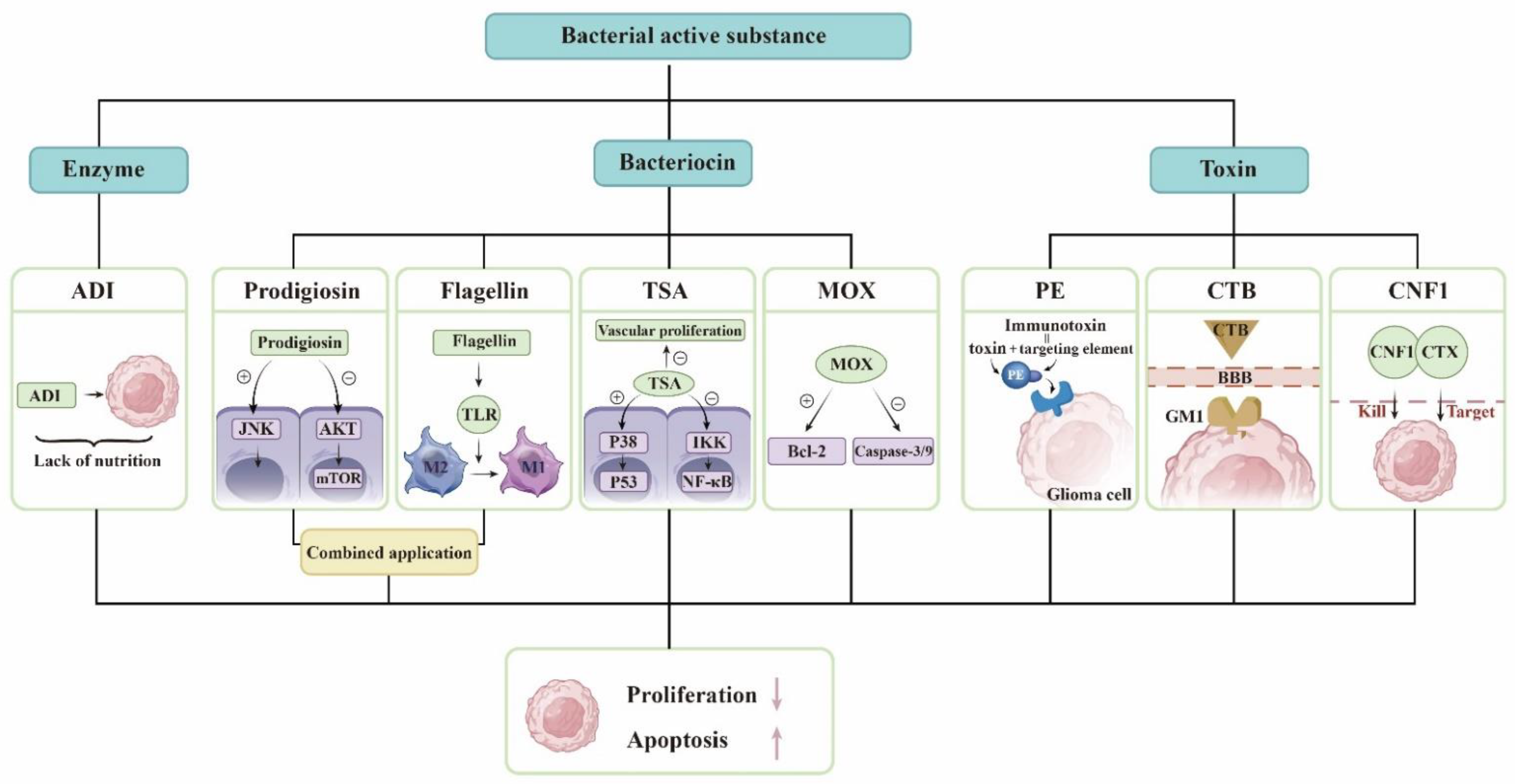
Figure 1. Derivatives of microorganisms are used to treat gliomas. The bacterial active substance includes enzymes, bacteriocins, and bacterial toxins. The list some of substances and their mechanisms of action. ADI exhausts arginine in the tumor and makes the tumor lack of nutrition. Prodigiosin activates the JNK pathway and reduce the AKT/mTOR pathway in GBM cells. Flagellin modulates the conversion of TAM from M2 to M1 type. TSA leads to p53 phosphorylation by activating p38MAPK and inhibits NF-κB activity by inducing IKK dephosphorylation. TSA also inhibits GBM vascular proliferation. MOX increases the ratio of Bcl-2-associated X protein/B-cell lymphoma 2 and activates the caspase-3/-9 cascade. PE and proteins that target gliomas constitute immunotoxins. CTB can pass the BBB and is targeted. CTX and CNF1 are responsible for identifying and killing glioma cells, respectively. They all promote glioma cell apoptosis and inhibit glioma proliferation.
2. Microbial-Targeted Therapy for Glioma
2.1. Potential of Bacteria
William Coley, an American surgeon, administered Streptococcus pyogenes to cancer patients in 1891, marking the first use of bacteria to treat cancer. As medicine has evolved, researchers have discovered many types of bacteria that can be used in tumor treatment. In theory, bacterial therapies have many advantages over conventional treatments, such as the ability to target tumors and actively penetrate tumor tissue, which may be related to the unique hypoxic microenvironment of tumor tissue and the immunosuppression caused by tumors [35,36][12][13]. Bacteria can, on the one hand, kill tumors through their own toxicity and, on the other hand, activate immune cells in the tumor microenvironment, enhancing the clearance of tumors by the immune system [37][14]. The BBB consists of several components, including tight junctions, vascular basement membranes, and astrocyte terminals that cover endothelial cells to form a physical barrier that functions as a filtering barrier for capillaries. As a filtering barrier, the BBB prevents most anticancer agents from penetrating the tumor and limits the therapeutic effect, which is one of the reasons for the poor prognosis of glioma [38][15]. Many bacteria can cross the blood–brain barrier and enter the center through a unique mechanism, which sets the stage for bacterial entry into targeted gliomas [39][16]. Bacterial therapy has been extensively studied in tumors of different tissues [40][17]. Theoretically, it seems that this therapy could also be used for gliomas, but there are still few studies on gliomas. The brain is more delicate than other tissues, and bacterial infection will have serious consequences. In the event of a brain abscess and brain hemorrhage, patients will be at risk of death [41][18]. How can the damage caused by bacteria to nontumor tissues be mitigated? How can the occurrence of bacteremia and shock be avoided?2.2. Oncolytic Virus
The concept of an oncolytic virus (OV) was first demonstrated in a case report in 1912 when DePace described a woman with cervical cancer who exhibited tumor regression after receiving an attenuated rabies virus vaccine [45][19]. With the development of viral genetic engineering techniques, the potential of lysing viruses was discovered. OVs have been more extensively studied in glioma than the bacteria mentioned above. Indeed, there are some features that make gliomas particularly suitable for lytic virus therapy. Gliomas are confined to the brain and lack distant metastasis, which favors the intratumor spread of OVs. The tumor growth is mainly surrounded by postmitotic cells, which facilitates virus replication with an active cell cycle [46][20]. There are multiple reasons for OVs targeting glioma. Some viruses have their own mechanisms or use immune cells as carriers that can cross the BBB to reach the site of glioma when administered systemically [47][21]. Receptors for some viruses can be highly expressed in tumor cells, such as the Poliovirus receptor CD155-targeted oncolysis of glioma [48][22]. Tumor tissue often exhibits viral immune defects. In normal tissues, the interferon pathway activates the downstream cascade signaling and activates immune cells, while tumor tissues are interferon-deficient [49][23]. Protein kinase R (PKR) is a strong inhibitor of viral protein synthesis and tumors, with activated Ras pathways showing impaired PKR function [46][20]. Tumor cells are replicatively and metabolically active, which also facilitates the replication of the virus. Similarly, OVs not only have a direct tumorolytic effect on cancer cells but also boost the immune system’s antitumor response. When infected cancer cells are lysed, they release additional infectious viral particles that aid in the destruction of the remaining tumor. OVs can induce glioblastoma autophagy and also break the immunosuppressed glioma microenvironment, activate the immune system, positively modulate immune synapses, and block immunosuppressive tumor metabolic circuits to inhibit tumor growth in different ways [50,51,52][24][25][26]. Viruses, such as herpes simplex virus-1, adenovirus, vaccinia virus, myxoma virus, and parvovirus, have been considered as glioma lysis agents [53][27]. Herpes simplex virus type 1 (HSV-1) is an enveloped double-stranded DNA virus known to infect and replicate in neural tissue, making it a potential treatment for glioma. Utilizing gene-editing techniques has resulted in a HSV with enhanced properties. γ34.5 is a viral antagonistic protein known to block protein kinase R (PKR) antiviral signaling in infected cells. HSV-1716 was obtained by deleting two copies of γ34.5/RL1 with weak toxicity. HSV-1 G207 is made safer by the insertion of the Escherichia coli lacZ gene into the coding sequence for the viral ICP6 gene and deletion of both copies of γ34.5 loci within the viral genome [53][27]. HSV-1716 and HSV-1 G207 have both been utilized in clinical research [54][28]. ICP47 supports HSV1 proliferation by reducing host cell-induced immune destruction. It was obtained by deleting the ICP34.5 and ICP47 from the appropriate locations to obtain talimogene laherparepvec, sold under the brand name Imlygic (T-VEC). T-VEC has passed phase III clinical trials and has been approved for clinical use by the FDA and the European Medicines Agency [54][28]. PVSRIPO, a chimera composed of poliovirus and rhinovirus, also completed the phase I clinical trial and received breakthrough treatment. Poliovirus has an affinity for CD155, which is highly expressed in gliomas and mediates its targeting to tumors. Intratumoral infusion of PVSRIPO causes the activation of antiviral responses and increases immune system activity, which provides an ideal platform for antitumor immunity [55][29]. Proinflammatory chemokines and neutrophils were increased in the tumor microenvironment treated with PVSRIPO. Neutrophils are able to produce TNF-α, induce nitric oxide synthase (iNOS), and also regulate the functions of NK, T, and B cells, which have long-lasting antitumor effects [56][30]. Parvovirus H-1 (H-1PV) is an oncolytic single-stranded DNA virus. The natural hosts of H-1PV are rodents, which have the ability to infect and replicate in humans, but lack pathogenicity. H-1PV interacts with galectin-1 on the cell surface and uses this glycoprotein to enter cancer cells [57][31], and its tumorolytic mechanism of action is thought to work through the cathepsin-mediated cell death pathway, so it may be an effective therapeutic approach for targeting glioma cells with defects in the apoptotic pathway [58][32]. It is able to cross the blood–brain barrier and therefore has potential for intravenous administration [59][33]. In phases I and II clinical studies, it met the primary objectives of safety and tolerability. There were no signs of systemic inflammation, excessive immune activation, or main organ toxicity [60][34]. In short, it is also a promising virus for targeting glioma. Treating gliomas with oncolytic bacteria also presents challenges, such as side effects. Common adverse effects include cerebral edema, hemiparesis, epilepsy, cerebral hemorrhage, aseptic meningitis, and fever [61][35]. Immune activation in the brain may cause inflammation, and excessive inflammation can trigger cerebral edema. In cases of more widespread brain edema, symptoms such as headache, altered mental status, nausea, and vomiting may occur. For example, in the clinical trial of PVSRIPO, patients developed cerebral hemorrhage and neurological symptoms [62][36]. A new direction in lysovirus therapy is a combination therapy, which combines the virus with other treatment strategies [63]. Combining OVs with antitumor drugs or immunostimulatory drugs can create synergistic effects and break drug resistance and immune tolerance [64,65]. OVs prevent the repair of tumor cell DNA and make the tumor more sensitive to radiation therapy [66] (Figure 2).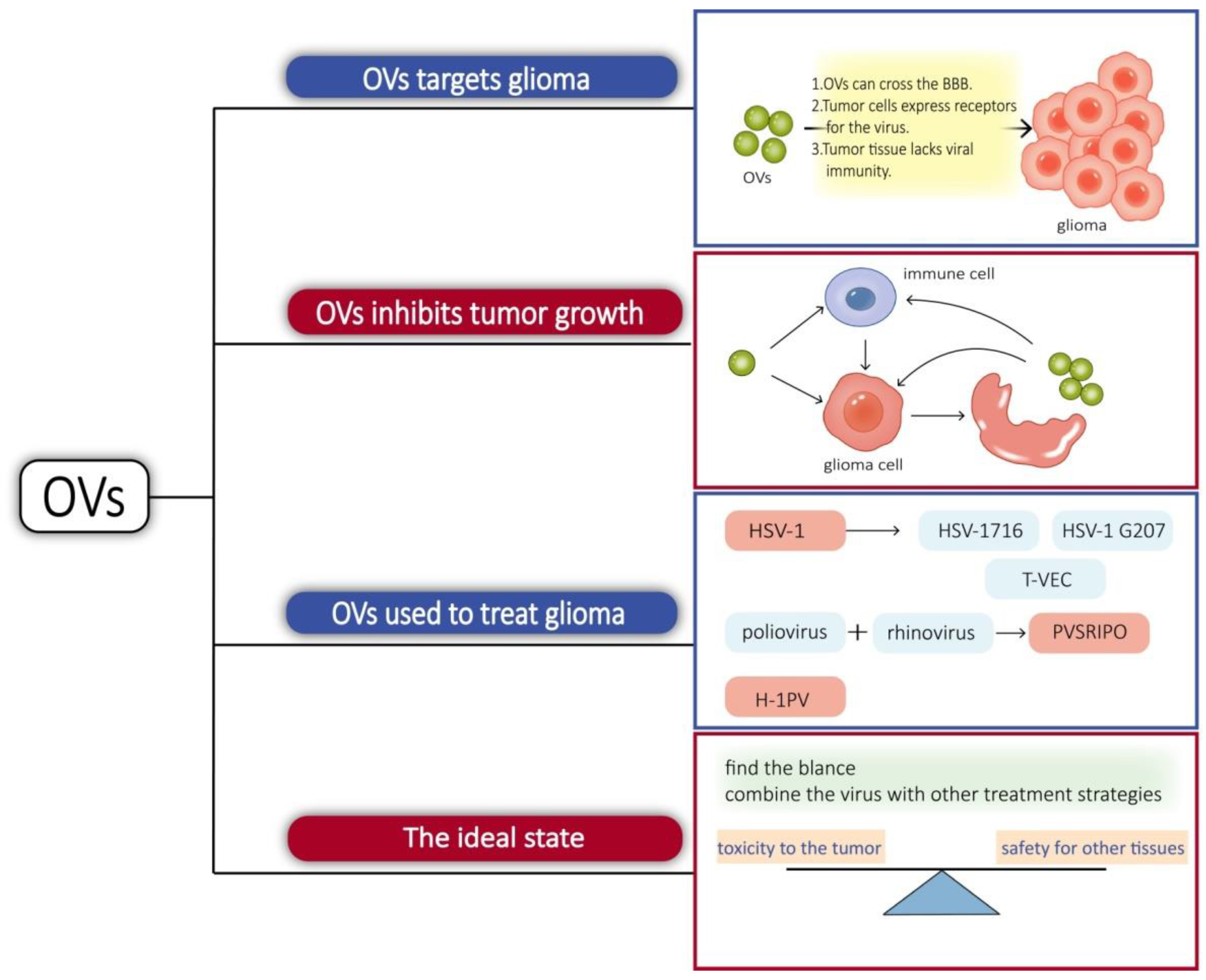
Figure 2. Some viruses can cross the BBB; some viruses can bind to receptors on the surface of glioma cells, and tumor tissue lacks immunity to viruses, so OVs tend to proliferate in glioma cells. On the one hand, OVs invade glioma cells, causing cells lysis and releasing new viruses that can invade other tumor cells, and, on the other hand, they can activate immune cells in tissues, breaking the immunosuppressive microenvironment of tumors and promoting immune cells to kill glioma cells.
3. Phages Can Target Gliomas for Drug Delivery
Phages are also effective carriers for glioma therapy, have a greater safety profile, do not normally proliferate in mammalian hosts [31][37], and can be employed to transport medications across the BBB [67][38]. Phage display technology involves inserting DNA sequences of exogenous peptides into phage shell protein genes so that both peptides and shell proteins are expressed on the phage surface. Using biopanning, targeting peptides or antibodies with a high affinity for tumors are selected, and these proteins can be used as drugs or drug carriers after modification [68][39]. GICP (glioma-initiating cell peptide), which exhibits a strong affinity for VAV3 protein, was identified using the phage display method [69][40]. Thus, it is possible to deliver medications precisely to gliomas using phages. One group investigated the efficacy of systemic temozolomide-activated phage-targeted gene therapy for GBM. The single-stranded genome of the human adeno-associated virus (AAV) was inserted into M13 phage, whose capsid was designed to display an RGD4C ligand, which binds to a αvβ3 integrin receptor, which is overexpressed on tumor cells, and once they bind, RGD4C/AAVP viral particles enter the cell, upon which the AAV genome is released to express genes from the cytomegalovirus CMV promoter. The CMV promoter is replaced with the Grp78 promoter, which can be activated by GBM or stimulated by temozolomide (TMZ) [70][41] (Figure 3).
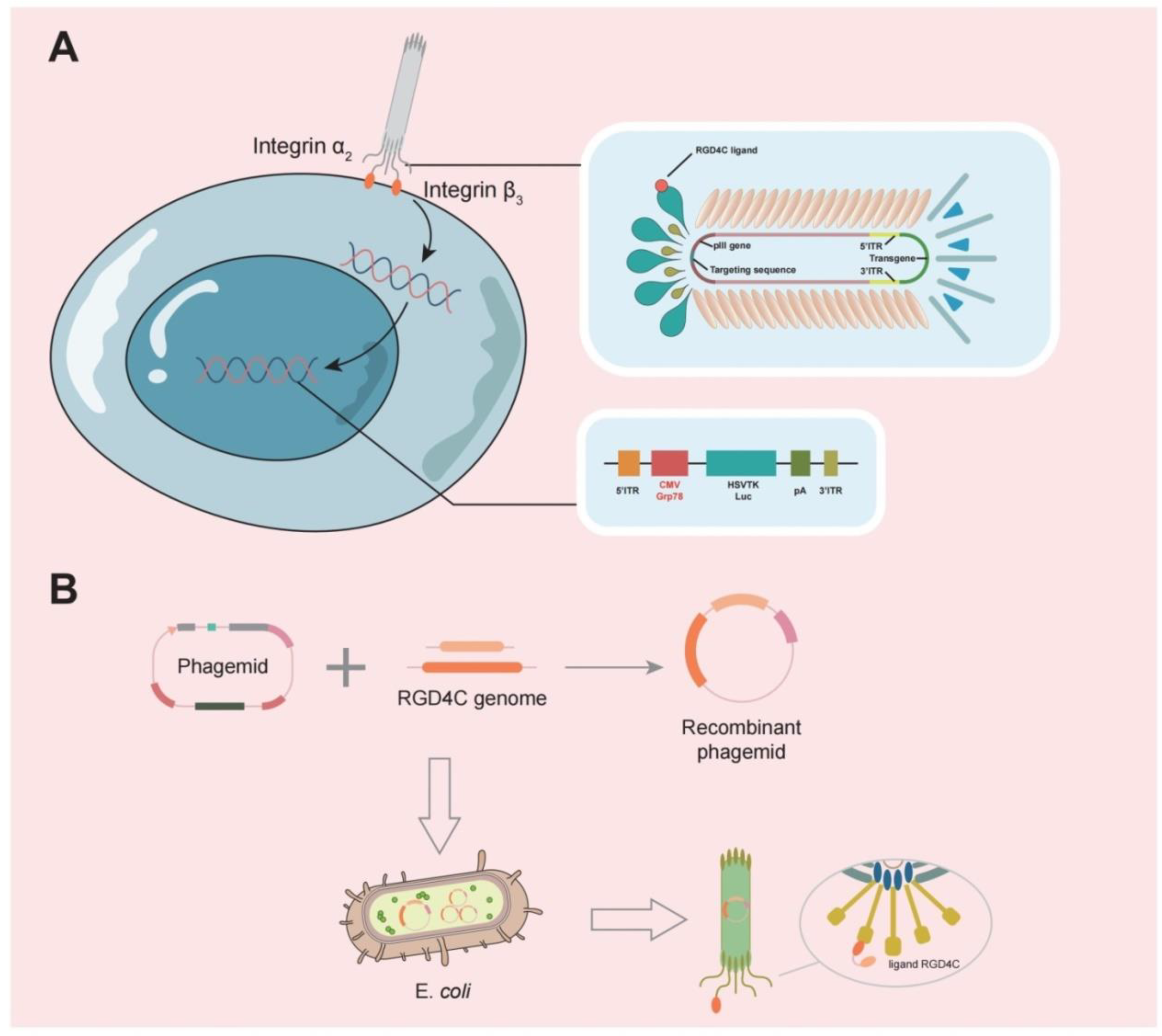
Figure 3. (A) A typical representation of M13 phage contains a single-stranded circular DNA, and the major coat proteins are pIII (green), pVIII (orange), and pVII + pIX complex (blue).The RGD4C ligand of this phage is individually designed to bind to the α2β3 integrin receptor, allowing it to bind to tumor cells and release the AAV genome into tumor cells. (B) The RGD4C ligand DNA sequence was inserted into the appropriate position of the phage shell protein structural gene by genetic engineering techniques, so that the RGD4C ligand gene is expressed along with the shell protein.
References
- Cheng, S.-Y.; Chen, N.-F.; Kuo, H.-M.; Yang, S.-N.; Sung, C.-S.; Sung, P.-J.; Wen, Z.-H.; Chen, W.-F. Prodigiosin stimulates endoplasmic reticulum stress and induces autophagic cell death in glioblastoma cells. Apoptosis 2018, 23, 314–328.
- Zheng, J.H.; Nguyen, V.H.; Jiang, S.-N.; Park, S.-H.; Tan, W.; Hong, S.H.; Shin, M.G.; Chung, I.-J.; Hong, Y.; Bom, H.-S.; et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 2017, 9, eaak9537.
- Hsu, Y.F.; Sheu, J.R.; Hsiao, G.; Lin, C.H.; Chang, T.H.; Chiu, P.T.; Wang, C.Y.; Hsu, M.J. p53 in trichostatin A induced C6 glioma cell death. Biochim. Biophys. Acta 2011, 1810, 504–513.
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747.
- Song, D.; Liang, H.; Qu, B.; Li, Y.; Liu, J.; Chen, C.; Zhang, D.; Zhang, X.; Gao, A. Moxidectin inhibits glioma cell viability by inducing G0/G1 cell cycle arrest and apoptosis. Oncol. Rep. 2018, 40, 1348–1358.
- Pang, Z.; Gu, M.-D.; Tang, T. Pseudomonas aeruginosa in Cancer Therapy: Current Knowledge, Challenges and Future Perspectives. Front. Oncol. 2022, 12, 891187.
- Guan, J.; Zhang, Z.; Hu, X.; Yang, Y.; Chai, Z.; Liu, X.; Liu, J.; Gao, B.; Lu, W.; Qian, J.; et al. Cholera Toxin Subunit B Enabled Multifunctional Glioma-Targeted Drug Delivery. Adv. Healthc. Mater. 2017, 6, 1700709.
- Tantillo, E.; Colistra, A.; Vannini, E.; Cerri, C.; Pancrazi, L.; Baroncelli, L.; Costa, M.; Caleo, M. Bacterial Toxins and Targeted Brain Therapy: New Insights from Cytotoxic Necrotizing Factor 1 (CNF1). Int. J. Mol. Sci. 2018, 19, 1632.
- Vannini, E.; Mori, E.; Tantillo, E.; Schmidt, G.; Caleo, M.; Costa, M. CTX-CNF1 Recombinant Protein Selectively Targets Glioma Cells In Vivo. Toxins 2021, 13, 194.
- Fiedler, T.; Strauss, M.; Hering, S.; Redanz, U.; William, D.; Rosche, Y.; Classen, C.F.; Kreikemeyer, B.; Linnebacher, M.; Maletzki, C. Arginine deprivation by arginine deiminase of Streptococcus pyogenes controls primary glioblastoma growth in vitro and in vivo. Cancer Biol. Ther. 2015, 16, 1047–1055.
- Hou, X.; Chen, S.; Zhang, P.; Guo, D.; Wang, B. Targeted Arginine Metabolism Therapy: A Dilemma in Glioma Treatment. Front. Oncol. 2022, 12, 938847.
- Leschner, S.; Westphal, K.; Dietrich, N.; Viegas, N.; Jablonska, J.; Lyszkiewicz, M.; Lienenklaus, S.; Falk, W.; Gekara, N.O.; Loessner, H.; et al. Tumor Invasion of Salmonella enterica Serovar Typhimurium Is Accompanied by Strong Hemorrhage Promoted by TNF-α. PLoS ONE 2009, 4, e6692.
- Zhou, S.; Gravekamp, C.; Bermudes, D.; Liu, K. Tumour-targeting bacteria engineered to fight cancer. Nat. Rev. Cancer 2018, 18, 727–743.
- Duong, M.T.-Q.; Qin, Y.; You, S.-H.; Min, J.-J. Bacteria-cancer interactions: Bacteria-based cancer therapy. Exp. Mol. Med. 2019, 51, 1–15.
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.-F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603.e4.
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial-Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell Mol. Neurobiol. 2018, 38, 1349–1368.
- Guo, Y.; Chen, Y.; Liu, X.; Min, J.-J.; Tan, W.; Zheng, J.H. Targeted cancer immunotherapy with genetically engineered oncolytic Salmonella typhimurium. Cancer Lett. 2020, 469, 102–110.
- Finnie, J.W.; Uzal, F.A. Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin. Int. J. Mol. Sci. 2022, 23, 9050.
- Rong, L.; Ni Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 1–18.
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: Concepts and candidates. Cancer J. 2012, 18, 69–81.
- Suryawanshi, Y.; Schulze, A. Oncolytic Viruses for Malignant Glioma: On the Verge of Success? Viruses 2021, 13, 1294.
- Merrill, M.K.; Bernhardt, G.; Sampson, J.; Wikstrand, C.J.; Bigner, D.D.; Gromeier, M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-Oncology 2004, 6, 208–217.
- Stojdl, D.F.; Lichty, B.D.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825.
- Kamynina, M.; Tskhovrebova, S.; Fares, J.; Timashev, P.; Laevskaya, A.; Ulasov, I. Oncolytic Virus-Induced Autophagy in Glioblastoma. Cancers 2021, 13, 3482.
- Achard, C.; Surendran, A.; Wedge, M.-E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24.
- Nguyen, T.T.; Shin, D.H.; Sohoni, S.; Singh, S.K.; Rivera-Molina, Y.; Jiang, H.; Fan, X.; Gumin, J.; Lang, F.F.; Alvarez-Breckenridge, C.; et al. Reshaping the tumor microenvironment with oncolytic viruses, positive regulation of the immune synapse, and blockade of the immunosuppressive oncometabolic circuitry. J. Immunother. Cancer 2022, 10, e004935.
- Rius-Rocabert, S.; García-Romero, N.; García, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic Virotherapy in Glioma Tumors. Int. J. Mol. Sci. 2020, 21, 7604.
- Carpenter, A.; Aiken, R.; Hanft, S. Oncolytic virus in gliomas: A review of human clinical investigations. Ann. Oncol. 2021, 32, 968–982.
- Gromeier, M.; Nair, S.K. Recombinant Poliovirus for Cancer Immunotherapy. Annu. Rev. Med. 2018, 69, 289–299.
- Holl, E.K.; Brown, M.C.; Boczkowski, D.; McNamara, M.A.; George, D.J.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget 2016, 7, 79828–79841.
- Ferreira, T.; Kulkarni, A.; Bretscher, C.; Nazarov, P.V.; Hossain, J.A.; Ystaas, L.A.R.; Miletic, H.; Röth, R.; Niesler, B.; Marchini, A. Oncolytic H-1 Parvovirus Hijacks Galectin-1 to Enter Cancer Cells. Viruses 2022, 14, 1018.
- Immidisetti, A.; Nwagwu, C.; Adamson, D.; Patel, N.; Carbonell, A.-M. Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults. Biomedicines 2021, 9, 138.
- Geletneky, K.; Leoni, A.-L.; Pohlmeyer-Esch, G.; Loebhard, S.; Leuchs, B.; Hoefer, C.; Jochims, K.; Dahm, M.; Huber, B.; Rommelaere, J.; et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp. Med. 2015, 65, 36–45.
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634.
- Shoaf, M.L.; Desjardins, A. Oncolytic Viral Therapy for Malignant Glioma and Their Application in Clinical Practice. Neurotherapeutics 2022, 1–14.
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161.
- Qi, Z.; Long, X.; Liu, J.; Cheng, P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front. Cell. Neurosci. 2022, 16, 819363.
- Kadhim, Z.A.; Sulaiman, G.M.; Al-Shammari, A.M.; Khan, R.A.; Al Rugaie, O.; Mohammed, H.A. Oncolytic Newcastle Disease Virus Co-Delivered with Modified PLGA Nanoparticles Encapsulating Temozolomide against Glioblastoma Cells: Developing an Effective Treatment Strategy. Molecules 2022, 27, 5757.
- Mahmoud, A.B.; Ajina, R.; Aref, S.; Darwish, M.; Alsayb, M.; Taher, M.; AlSharif, S.A.; Hashem, A.M.; Alkayyal, A.A. Advances in immunotherapy for glioblastoma multiforme. Front. Immunol. 2022, 13, 944452.
- Asija, S.; Chatterjee, A.; Yadav, S.; Chekuri, G.; Karulkar, A.; Jaiswal, A.K.; Goda, J.S.; Purwar, R. Combinatorial approaches to effective therapy in glioblastoma (GBM): Current status and what the future holds. Int. Rev. Immunol. 2022, 41, 582–605.
- Wang, Y.; Sheng, J.; Chai, J.; Zhu, C.; Li, X.; Yang, W.; Cui, R.; Ge, T. Filamentous Bacteriophage—A Powerful Carrier for Glioma Therapy. Front. Immunol. 2021, 12, 729336.
- Wu, L.-P.; Ahmadvand, D.; Su, J.; Hall, A.; Tan, X.; Farhangrazi, Z.S.; Moghimi, S.M. Crossing the blood-brain-barrier with nanoligand drug carriers self-assembled from a phage display peptide. Nat. Commun. 2019, 10, 1–16.
- Saw, P.E.; Song, E.-W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807.
- Zhang, M.; Lu, W. Enhanced glioma-targeting and stability of L GICP peptide coupled with stabilized peptide D A7R. Acta Pharm. Sin. B 2018, 8, 106–115.
- Przystal, J.M.; Waramit, S.; Pranjol, Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M.; et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492.
More