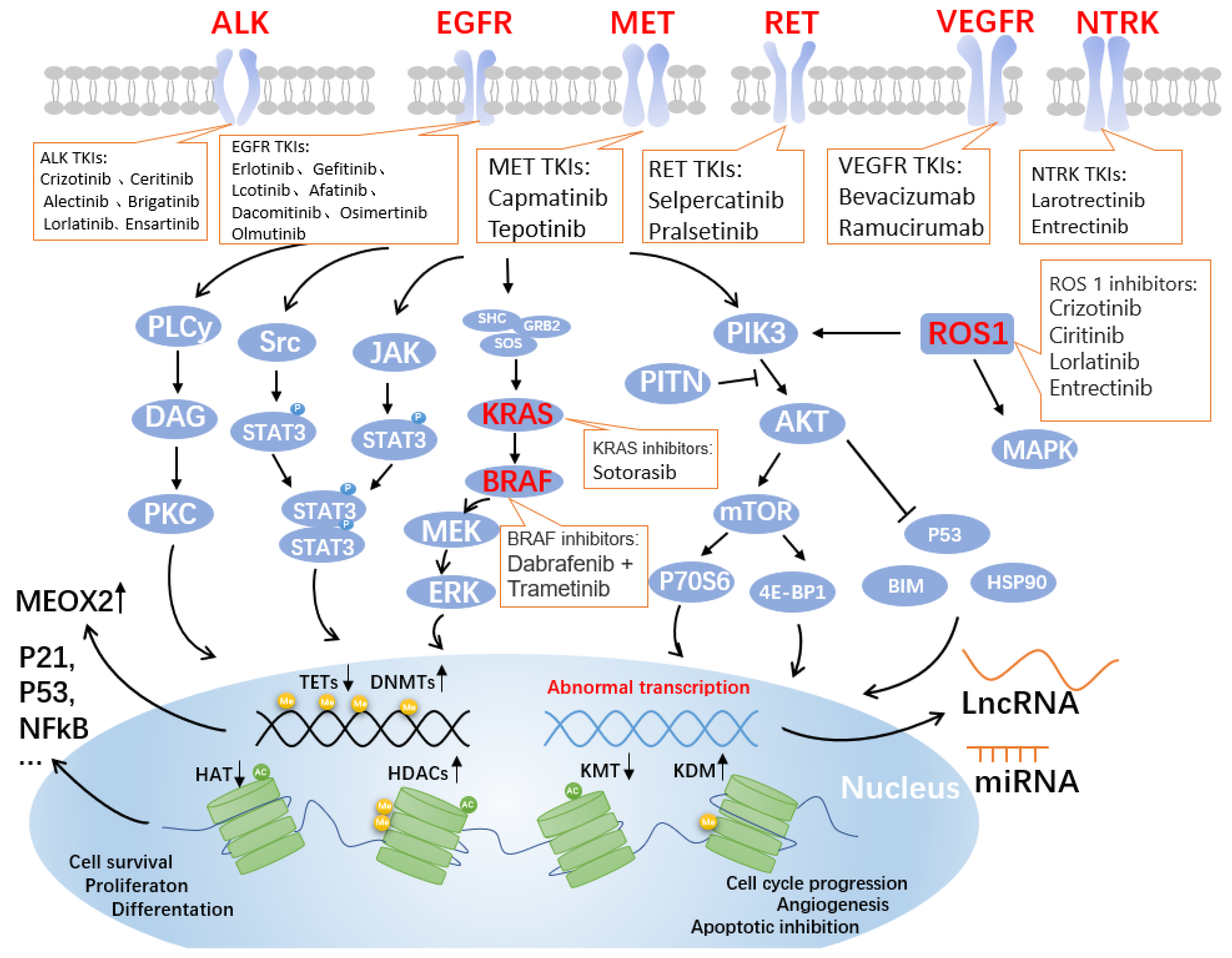
The advent of precision medicine has brought light to the treatment of non-small cell lung cancer (NSCLC), expanding the options for patients with advanced NSCLC by targeting therapy through genetic and epigenetic cues. Tumor driver genes in NSCLC patients have been uncovered one by one, including epidermal growth factor receptor (EGFR), mesenchymal lymphoma kinase (ALK), and receptor tyrosine kinase ROS proto-oncogene 1 (ROS1) mutants. Antibodies and inhibitors that target the critical gene-mediated signaling pathways that regulate tumor growth and development are anticipated to increase patient survival and quality of life. Targeted drugs continue to emerge, with as many as two dozen approved by the FDA, and chemotherapy and targeted therapy have significantly improved patient prognosis.
- NSCLC
- EGFR
- drug
1. EGFR-TKIs
| Generation | Drug | Approval Status | Reversible/Irreversible | Median PFS (Months) |
Ref. |
|---|---|---|---|---|---|
| 1st | Erlotinib | FDA, EMA | Reversible | 9.7 | [13][4] |
| Gefitinib | FDA, EMA | Reversible | 10.8 | [14][5] | |
| Icotinib | CFDA | Reversible | 10 | [18][9] | |
| 2nd | Afatinib | FDA, EMA, CFDA | Irreversible | 11.0 | [19][10] |
| Dacomitinib | FDA | Irreversible | 14.7 | [20][11] | |
| 3rd | Osimertinib | FDA, MEA | Irreversible | 18.9 | [21][12] |
| Olmutinib | KFDA (Conditional) | Irreversible | NR | [19][10] |
2. ALK-TKIs
| Generation | Drug | Objective Response Rate (ORR) |
Median PFS (Months) |
Side Effects | Ref. |
|---|---|---|---|---|---|
| 1st | Crizotinib | 74% | 10.9 | Vision disorder/ nausea/diarrhea |
[31][23] |
| 2nd | Ceritinib | 73% | 16.6 | Diarrhea/nausea vomiting |
[32,33][24][25] |
| Alectinib | 83% | 25.7 | AST elevation/CK elevation/fatigue |
[32,33][24][25] | |
| Brigatinib | 74% | 24 | nausea/diarrhea/cough | [32,33][24][25] | |
| 3rd | Lorlatinib | 76% | NR | Hypercholesterolemia/edema/peripheral neuropathy/ | [33][25] |
| Ensartinib | 75% | 25.8 | rash/ALT elevation/AST elevation | [33,34][25][26] |
3. Other Targeted Sites and Drugs for NSCLC
3. Other Targeted Sites and Drugs for Non-Small Cell Lung Cancer
3.1. ROS1
3.2. BRAF
3.3. MET
3.4. RET
3.5. KRAS
3.6. VEGF
| Targeted Genes | Drug | Objective Response Rate (ORR) | Median PFS (Months) |
Side Effects | Ref. |
|---|---|---|---|---|---|
| ROS1 | Crizotinib | 72.4% | 19.2 | Visual impairment/nausea/edema/ | [35,76][27][68] |
| Ciritinib | 62% (67%) * | 19.3 | diarrhea/nausea/anorexia/ | [43][35] | |
| Lorlatinib | 41% (62%) * | 8.5 | dyslipidemia | [43][35] | |
| Entrectinib | 77% | 15.7 | weight increase/neutropenia | [77,78][69][70] | |
| BRAF | Dabrafenib and trametinib | 64% (68%) * | 10.8 | Fatigue/pyrexia/nausea | [79][71] |
| MET | Tepotinib | 46% | 8.5 | Peripheral edema/amylase increased/nausea | [80][72] |
| Capmatinib | 41% (68%) | 5.4 | peripheral edema/Nausea | [81][73] | |
| RET | Selpercatinib | 64% (85%) | 18.4 | Dry mouth/diarrhea/hypertension | [82,[7483]][75] |
| Pralsetinib | 61% (73%) * | 16.5 (13) * | anemia/hypertension/neutropenia/ | [83][75] | |
| KRAS | Sotorasib | 32% | 6.3 | diarrhea/nausea/elevated LFT/fatigue | [71][63] |
| NTRK | Larotrectinib | 75% | 35.4 | myalgia/hypersensitivity/weight increase | [84][76] |
| Entrectinib | 70% | NR | taste disorder/ constipation/fatigue |
[84][76] | |
| HER2 | T-DM1 | 55% | 5 | Infusion reactions/ thrombocytopenia |
[85,86][77][78] |
| T-DXd | 62% | 14 | nausea /alopecia/anemia |
[87][79] |
References
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 53.
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246.
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19.
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388.
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; Obyrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334.
- Remon, J.; Steuer, C.; Ramalingam, S.; Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 2018, 29, i20–i27.
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125.
- Liang, J.L.; Ren, X.C.; Lin, Q. Treating advanced non-small-cell lung cancer in Chinese patients: Focus on icotinib. Onco. Targets Ther. 2014, 7, 761–770.
- Juan, O.; Popat, S. Treatment choice in epidermal growth factor receptor mutation-positive non-small cell lung carcinoma: Latest evidence and clinical implications. Ther. Adv. Med. Oncol. 2017, 9, 201–216.
- Lavacchi, D.; Mazzoni, F.; Giaccone, G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): Current perspectives. Drug Des. Devel. Ther. 2019, 13, 3187–3198.
- Le, T.; Gerber, D.E. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers 2019, 11, 366.
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164.
- Duyster, J.; Bai, R.-Y.; Morris, S.W. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001, 20, 5623–5637.
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19.
- Zhang, S.S.; Nagasaka, M.; Zhu, V.W.; Ou, S.-H.I. Going beneath the tip of the iceberg. Identifying and understanding EML4-ALK variants and TP53 mutations to optimize treatment of ALK fusion positive (ALK+) NSCLC. Lung Cancer 2021, 158, 126–136.
- Sullivan, I.; Planchard, D. ALK inhibitors in non-small cell lung cancer: The latest evidence and developments. Ther. Adv. Med. Oncol. 2015, 8, 32–47.
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401.
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703.
- Markham, A. Brigatinib: First Global Approval. Drugs 2017, 77, 1131–1135.
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690.
- Karachaliou, N.; Santarpia, M.; Cao, M.G.; Teixido, C.; Sosa, A.E.; Berenguer, J.; Capote, A.R.; Altavilla, G.; Rosell, R. Anaplastic lymphoma kinase inhibitors in phase I and phase II clinical trials for non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 713–722.
- Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S.; Sahu, A. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97.
- Xia, B.; Nagasaka, M.; Zhu, V.W.; Ou, S.-H.I.; Soo, R.A. How to select the best upfront therapy for metastatic disease? Focus on ALK-rearranged non-small cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2020, 9, 2521–2534.
- Wang, Y.; Yuan, X.; Xiong, J.; Hao, Z.; Peng, X.; Chen, W.; Cui, L.; Li, H.; Wang, X.; He, X.; et al. Pharmacology and Clinical Evaluation of Ensartinib Hydrochloride Capsule. Zhongguo Fei Ai Za Zhi 2020, 23, 719–729.
- Horn, L.; Wang, Z.; Wu, G.; Poddubskaya, E.; Mok, T.; Reck, M.; Wakelee, H.; Chiappori, A.A.; Lee, D.H.; Breder, V.; et al. Ensartinib vs. Crizotinib for Patients With Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1617–1625.
- D’Angelo, A.; Sobhani, N.; Chapman, R.; Bagby, S.; Bortoletti, C.; Traversini, M.; Ferrari, K.; Voltolini, L.; Darlow, J.; Roviello, G. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers 2020, 12, 3293.
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. Biophys. Acta 2009, 1795, 37–52.
- Bergethon, K.; Shaw, A.T.; Ou, S.-H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870.
- Guaitoli, G.; Bertolini, F.; Bettelli, S.; Manfredini, S.; Maur, M.; Trudu, L.; Aramini, B.; Masciale, V.; Grisendi, G.; Dominici, M.; et al. Deepening the Knowledge of ROS1 Rearrangements in Non-Small Cell Lung Cancer: Diagnosis, Treatment, Resistance and Concomitant Alterations. Int. J. Mol. Sci. 2021, 22, 12867.
- Patil, T.; Simons, E.; Mushtaq, R.; Pacheco, J.; Doebele, R.; Bowles, D. Targeted therapies for ROS1-rearranged non-small cell lung cancer. Drugs Today 2019, 55, 641–652.
- De Giglio, A.; Lamberti, G.; Facchinetti, F.; Genova, C.; Andrini, E.; Bello, M.G.D.; Tiseo, M.; Metro, G.; Chiari, R.; Ricciuti, B. Treatment Patterns and Clinical Outcomes Among Patients With ROS1-rearranged Non-small-cell Lung Cancer Progressing on Crizotinib. Clin. Lung Cancer 2020, 21, e478–e487.
- Lin, J.J.; Shaw, A.T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. 2017, 12, 1611–1625.
- Dziadziuszko, R.; Le, A.T.; Wrona, A.; Jassem, J.; Camidge, D.R.; Varella-Garcia, M.; Aisner, D.L.; Doebele, R.C. An Activating KIT Mutation Induces Crizotinib Resistance in ROS1-Positive Lung Cancer. J. Thorac. Oncol. 2016, 11, 1273–1281.
- Azelby, C.M.; Sakamoto, M.R.; Bowles, D.W. ROS1 Targeted Therapies: Current Status. Curr. Oncol. Rep. 2021, 23, 94.
- Negrao, M.V.; Raymond, V.M.; Lanman, R.B.; Robichaux, J.P.; He, J.; Nilsson, M.B.; Ng, P.K.; Amador, B.E.; Roarty, E.B.; Nagy, R.J.; et al. Molecular Landscape of BRAF-Mutant NSCLC Reveals an Association Between Clonality and Driver Mutations and Identifies Targetable Non-V600 Driver Mutations. J. Thorac. Oncol. 2020, 15, 1611–1623.
- Loo, E.; Khalili, P.; Beuhler, K.; Siddiqi, I.; Vasef, M.A. BRAF V600E Mutation Across Multiple Tumor Types: Correlation Between DNA-based Sequencing and Mutation-specific Immunohistochemistry. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 709–713.
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Aragaki, J.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; et al. Different effects of point mutations within the B-Raf glycine-rich loop in colorectal tumors on mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase and nuclear factor kappaB pathway and cellular transformation. Cancer Res. 2004, 64, 3428–3435.
- Marchetti, A.; Felicioni, L.; Malatesta, S.; Sciarrotta, M.G.; Guetti, L.; Chella, A.; Viola, P.; Pullara, C.; Mucilli, F.; Buttitta, F. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 3574–3579.
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 2018, 23, 740–745.
- O’Leary, C.G.; Andelkovic, V.; Ladwa, R.; Pavlakis, N.; Zhou, C.; Hirsch, F.; Richard, D.; O’Byrne, K. Targeting BRAF mutations in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 1119–1124.
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.S.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAF(V600) and BRAF(nonV600) mutations. Ann. Oncol. 2020, 31, 289–294.
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262.
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Woude, G.F.V. Mechanism of met oncogene activation. Cell 1986, 45, 895–904.
- Santarpia, M.; Massafra, M.; Gebbia, V.; D’Aquino, A.; Garipoli, C.; Altavilla, G.; Rosell, R. A narrative review of MET inhibitors in non-small cell lung cancer with MET exon 14 skipping mutations. Transl. Lung Cancer Res. 2021, 10, 1536–1556.
- Skead, G.; Govender, D. Gene of the month: MET. J. Clin. Pathol. 2015, 68, 405–409.
- Drilon, A.; Cappuzzo, F.; Ou, S.-H.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26.
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281.
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730.
- Han, S.; Ma, X.; Fang, J. Progress on Mechanism of MET Gene Mutation and Targeted Drugs in Non-small Cell Lung Cancer. Zhongguo Fei Ai Za Zhi 2020, 23, 609–614.
- Fujino, T.; Suda, K.; Mitsudomi, T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin. Emerg. Drugs 2020, 25, 229–249.
- Mathieu, L.N.; Larkins, E.; Akinboro, O.; Roy, P.; Amatya, A.K.; Fiero, M.H.; Mishra-Kalyani, P.S.; Helms, W.S.; Myers, C.E.; Skinner, A.M.; et al. FDA Approval Summary: Capmatinib and Tepotinib for the Treatment of Metastatic NSCLC Harboring MET Exon 14 Skipping Mutations or Alterations. Clin. Cancer Res. 2022, 28, 249–254.
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2017, 15, 151–167.
- Bronte, G.; Ulivi, P.; Verlicchi, A.; Cravero, P.; Delmonte, A.; Crinò, L. Targeting RET-rearranged non-small-cell lung cancer: Future prospects. Lung Cancer 2019, 10, 27–36.
- Ferrara, R.; Auger, N.; Auclin, E.; Besse, B. Clinical and Translational Implications of RET Rearrangements in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 27–45.
- Wright, K.M. FDA Approves Pralsetinib for Treatment of Adults With Metastatic RET Fusion-Positive NSCLC. Oncology 2020, 34, 406–431.
- Nguyen, L.; Monestime, S. Pralsetinib: Treatment of metastatic RET fusion-positive non-small cell lung cancer. Am. J. Health Syst. Pharm. 2022, 79, 527–533.
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879.
- Reck, M.; Carbone, D.; Garassino, M.; Barlesi, F. Targeting KRAS in non-small-cell lung cancer: Recent progress and new approaches. Ann. Oncol. 2021, 32, 1101–1110.
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223.
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579.
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381.
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217.
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2021, 28, 1482–1486.
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467.
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC. J. Thorac. Oncol. 2021, 16, 205–215.
- Hafner, S. First-line anti-VEGF plus EGFR-TKI in EGFR-mutant NSCLC: Adding the ARTEMIS trial to the puzzle of current evidence. Signal Transduct. Target. Ther. 2021, 6, 417.
- Shaw, A.; Riely, G.; Bang, Y.-J.; Kim, D.-W.; Camidge, D.; Solomon, B.; Varella-Garcia, M.; Iafrate, A.; Shapiro, G.; Usari, T.; et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126.
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.-J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2019, 21, 261–270.
- Dziadziuszko, R.; Krebs, M.G.; De Braud, F.; Siena, S.; Drilon, A.; Doebele, R.C.; Patel, M.R.; Cho, B.C.; Liu, S.V.; Ahn, M.-J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Locally Advanced or Metastatic ROS1 Fusion-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 1253–1263.
- Auliac, J.-B.; Bayle, S.; Do, P.; Le Garff, G.; Roa, M.; Falchero, L.; Huchot, E.; Quéré, G.; Jeannin, G.; Métivier, A.-C.; et al. Efficacy of Dabrafenib Plus Trametinib Combination in Patients with BRAF V600E-Mutant NSCLC in Real-World Setting: GFPC 01-2019. Cancers 2020, 12, 3608.
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943.
- Wu, Y.-L.; Smit, E.F.; Bauer, T.M. Capmatinib for patients with non-small cell lung cancer with MET exon 14 skipping mutations: A review of preclinical and clinical studies. Cancer Treat. Rev. 2021, 95, 102173.
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824.
- Cascetta, P.; Sforza, V.; Manzo, A.; Carillio, G.; Palumbo, G.; Esposito, G.; Montanino, A.; Costanzo, R.; Sandomenico, C.; De Cecio, R.; et al. RET Inhibitors in Non-Small-Cell Lung Cancer. Cancers 2021, 13, 4415.
- Qin, H.; Patel, M.R. The Challenge and Opportunity of NTRK Inhibitors in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 2916.
- Riudavets, M.; Sullivan, I.; Abdayem, P.; Planchard, D. Targeting HER2 in non-small-cell lung cancer (NSCLC): A glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open 2021, 6, 100260.
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J. Clin. Oncol. 2018, 36, 2532–2537.
- Azar, I.; Alkassis, S.; Fukui, J.; Alsawah, F.; Fedak, K.; Al Hallak, M.N.; Sukari, A.; Nagasaka, M. Spotlight on Trastuzumab Deruxtecan (DS-8201,T-DXd) for HER2 Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer 2021, 12, 103–114.