Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Ping-Heng Tan.
Interferons (IFNs) are pleiotropic cytokines originally identified for their antiviral activity. IFN-α and IFN-β are both type I IFNs that have been used to treat neurological diseases such as multiple sclerosis. Microglia, astrocytes, as well as neurons in the central and peripheral nervous systems, including spinal cord neurons and dorsal root ganglion neurons, express type I IFN receptors (IFNARs). Type I IFNs play an active role in regulating cognition, aging, depression, and neurodegenerative diseases. Notably, by suppressing neuronal activity and synaptic transmission, IFN-α and IFN-β produced potent analgesia.
- neuroinflammation
- neurological disease
- microglia
- astrocytes
- long-haul COVID
- IFN-a
- IFN-b
- spinal cord
1. Introduction
Interferons (IFNs) were first found in 1957 [1] and were found to be able to “interfere” with viruses [2]. In addition to antiviral effects, IFNs also could affect the function of the immune system, endocrine system, and nervous system, especially the central nervous system (CNS). As a whole, the IFN family can be divided into three subfamilies: the type I IFNs (IFN-Is), the type II IFN (which contains IFN-γ), and the type III IFNs (which contain IFN-λ1-3) [1,3][1][3]. IFN-Is include IFN-α (13 homologous human and 14 homologous mouse subtypes), IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-τ, and IFN-ω1–3. IFN-α, IFN-β, IFN-ε, IFN-τ, IFN-κ, and IFN-ω are human IFNs [4]. ThiRes review article willeawrchers focused on IFN-α and IFN-β. IFN-I family members are pleiotropic cytokines, and they are potent immunomodulatory factors that bridge the innate and adaptive immune responses and act as antimicrobial, antitumor, and pain mediators in the host [4,5,6][4][5][6].
During viral or bacterial infection and tissue injury, cytokines and chemokines, including IFN-Is, are released through activation of pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) that sense pathogen/damage-associated molecular patterns (PAMPs/DAMPs) through neuro-immune interactions [7,8][7][8]. TLR 2, 4, and 5, localized on outer membrane, respond primarily to bacterial surface associated PAMPs. TLR4 can also be found to a lesser extent in endosomes. TLR3 and TLR7/8, localized on endosomes, mainly respond to nucleic-acid-based PAMPs from viruses and bacteria such as double-stranded RNAs (dsRNA) and single-stranded RNAs (ssRNA), respectively [9,10][9][10]. TLR3 recognizes viral dsRNA and its synthetic analog, polyinosine-deoxycytidylic acid (poly(I:C)). Imiquimod and Resiquimod are imidazoquinoline-like molecules that have been identified as TLR7/8 agonists. TLR9 is localized on endosomes and senses double-stranded DNA (dsDNA) and CpG unmethylated DNA. IFN-I is induced by TLR4 upon recognition of lipopolysaccharides (LPS) and viral proteins from Gram-negative bacteria [7]. Activation of TLRs can produce large amounts of IFN-I and induce subsequent specific intracellular signaling pathways after sensing PAMPs and DAMPs. The ligand binding to the TLR initiates recruitment to the receptor of adaptor proteins TRIF (for TLR3) and MyD88 (for TLR7/8/9). In turn, this initiates a cytoplasmic signaling cascade that leads to the phosphorylation of IFN regulatory factors (IRFs) 3 and 7 by TANK binding kinase (TBK) and the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). Transcription of IFN-I genes is then mediated by IRFs binding to promoter/enhancer regions [11] (Figure 1).
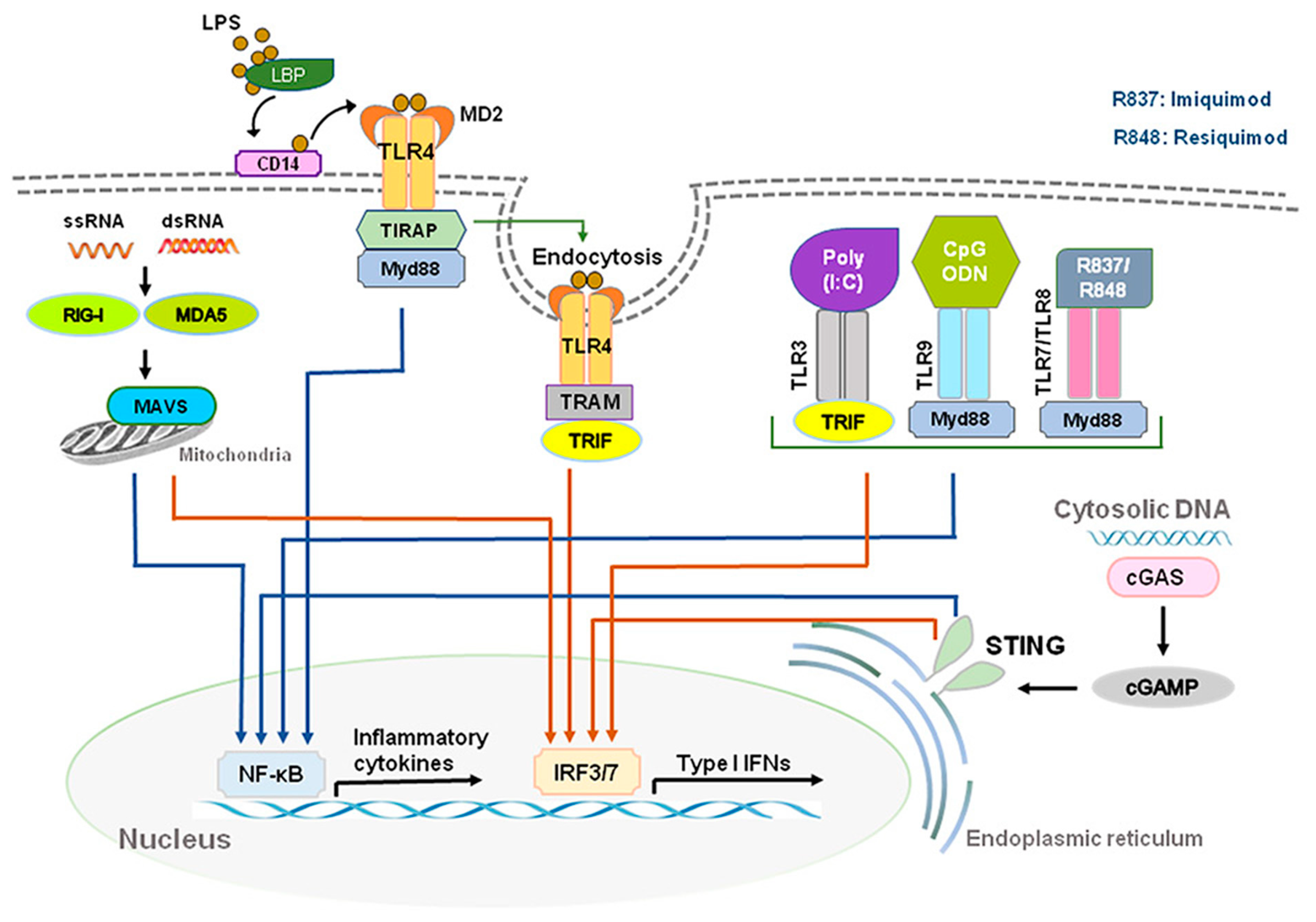
Figure 1. Production of type I IFNs. Type I IFNs are induced by activation of toll-like receptors (TLRs) and stimulator of interferon genes (STING). Abbreviations: CD14, cluster of differentiation 14; LBP, LPS-binding protein; LPS, lipopolysaccharide; MD2, myeloid differentiation factor 2; poly(I:C), polyinosine-deoxycytidylic acid; poly(I:C), polyinosine-deoxycytidylic acid; IRF3/7, interferon regulatory factor 3/7; MYD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; STING, stimulator of interferon genes; TRIF, TIR-domain-containing adapter-inducing IFN-β.
DExD/H-box RNA helicases and stimulator of interferon genes (STING) situated in the cytoplasm are also capable of detecting microbial RNA and DNA and producing IFN-I, in addition to TLRs [6]. The family includes three type of receptors, such as the retinoic acid inducible gene-1(RIG-I)-like helicases (RLHs), RIG-I, and melanoma differentiation-associated protein 5 (MDA5) [2,11][2][11]. STING is a protein with four putative transmembrane domains and resides in the endoplasmic reticulum (ER) and activates intracellular DNA-mediated production of IFN-I [9,12,13][9][12][13] (Figure 1). STING can be activated after binding cyclic dinucleotides (CDNs) derived from virus or intracellular DNA and activates the kinase TBK1 to induce phosphorylation of STING. Phosphorylated STING is coupled with TBK1 and recruits IRF3 to enter the nucleus, activating the production of IFN-I and promoting the eradication of pathogens mediated by immune cells [12,14][12][14]. Different types of cells could produce IFN-α and IFN-β, including macrophages, natural killer (NK) cells, fibroblasts, B cells, T cells, and osteoblasts. IFN-α and IFN-β can exert anti-viral and anti-tumor effects by stimulating NK cells and macrophages. During virus infection or stimulation with DNAs/RNAs, IFN-I is mainly produced and secreted by the plasmacytoid dendritic cells (pDC) [15,16,17][15][16][17].
2. Intracellular Signaling of IFN-I
IFN-Is bind to heterodimer interferon receptors (IFNAR1 and IFNAR2) [4,18,19,20,21][4][18][19][20][21] and subsequently recruit Janus family kinase1 (Jak1) and tyrosine kinase 2 (Tyk2) to phosphorylate and activate IFNAR1 and IFNAR2. JAKs are composed of the four family members of JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). IFNAR1 is coupled with TYK2, whereas IFNAR2 is coupled with JAK1 (Figure 2). In humans, the genes that code for IFN-Is are located on chromosome 9; these genes are located on chromosome 4 in mice [22]. IFNAR1 signaling is dependent on TYK2; IFNARII signaling is dependent on JAK1 (Figure 2). After receptor subunits rearrange and dimerize in response to ligand, these receptor-associated JAKs autophosphorylate to activate STAT (signal transducer and activator of transcription) [23]. IFNARs are activated and subsequently phosphorylate effector proteins of the signal transducers and activators of transcription (STAT) family [4]. Phosphorylated STAT1 and STAT2 couple with IRF9 to form the transcription factor ISGF3 and translocate to the nucleus to stimulate the transcription of IFN-stimulated genes (ISG), including antiviral genes and type I IFNs themselves [17,18][17][18] (Figure 2). Antiviral genes include ISG 15, 2′-5′ oligoadenylate synthetase (OAS), ribonuclease L (RNase L), orthomyxovirus resistance gene (Mx), and viperin, etc. Many of the biological effects of IFN-Is appear to be mediated by the activation of JAK-STAT signaling pathways. Activation of IFNAR1/2 also results in non-canonical signaling such as the activation of PI3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways [6] in addition to canonical signaling pathway (Figure 2).
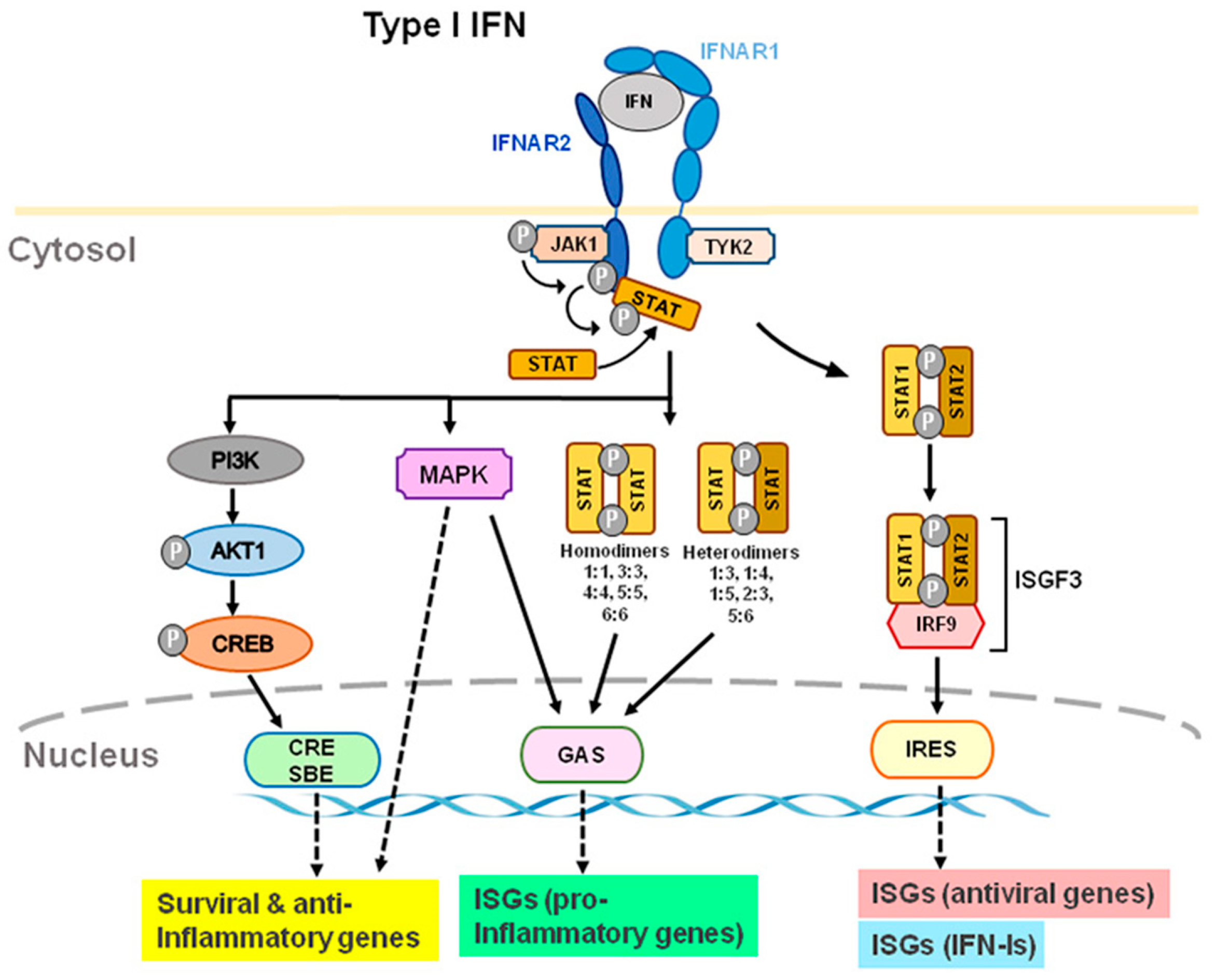
Figure 2. Type I IFN-induced interferon receptor (IFNAR) signaling and gene expression. IFN-α and IFN-β transmit signals via IFNAR1 and IFNAR2 subunits. TYK2 and JAK1 are distinct components of IFNAR1 and IFNAR2. Through ISRE, GAS, and CRE/SBE, TYK2 and JAK1 activation lead to activation of STAT1/2, STAT homo- or heterodimers, PI3K, and MAPK, which activates transcription of ISGs, including antiviral, type-I IFN, and pro-inflammatory genes. Furthermore, TYK2 may form associations with membrane proteins, such as ion channels, which modulate the activity of cells rapidly. Abbreviations: JAK1, Janus kinase 1; TYK2, tyrosine kinase 2; STAT, signal transducer and activator of transcription; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; CRE, cyclic AMP response element; SBE, SMAD binding elements; GAS, IFN-γ-activated sites; ISRE, IFN-stimulated response elements; ISG, IFN-stimulated genes.
Upon activating IFNRs and downstream signaling, IFN-I could induce multiple antiviral genes to inhibit virus replication in infected cells and prevent infection of nearby cells [3]. The suppressor of cytokine signaling (SOCS)1 is a potent inhibitor of JAK2 and could reduce IFN-Is response. Production of suppressor of SOCS1 expression is maintained by IFN-β or high amounts of IFNɑ2 [24]. SOCS-1 also enhances immunological actions of IFN-γ but inhibits the adverse effect of unregulated IFN-γ responses by inhibiting STAT1ɑ and decreasing the duration of IFN-γ signaling. By either binding directly to receptors or inhibiting JAK1, JAK2, and TYK2 directly, SOCS3 inhibits the receptor signaling but not JAK3 signaling. In comparison to SOCS1, SOCS3 is less potent than SOCS1 in suppressing IFN-γ signaling. By suppressing STAT3 signaling, SOCS3 also inhibits Th17 differentiation. Through IL-6 signaling, SOCS5 negatively regulates STAT6, an essential signaling molecule for IL-4, which inhibits Th2 differentiation [20].
Many types of viral infections, including hepatitis C and hepatitis B, have been successfully treated with IFN-Is. Multisystem autoimmune diseases, such as multiple sclerosis, are also treated with IFN-Is [25]. IFN-Is also were used to treat cancer through directly activating cytotoxic T lymphocytes, NK cell activation, induction of tumor cell death, and inhibition of angiogenesis. Treatment for melanoma with IFN-Is is reported to effectively improve disease-free survival [26]. IFN-Is can interact with host cells to induce protective immunity; e.g., IFN-I can enhance antibody production by dendritic cells. IFN-Is were also immunosuppressants and anti-inflammatory mediators by induction of programmed cell death-ligand 1 (PD-L1) [27] and anti-inflammatory cytokines (e.g., IL-10) [14] as well as blocking the expression of pro-inflammatory mediators, such as matrix metalloproteinase 9 (MMP-9), ICAM-1, VCAM-1, and tumor necrosis factor-α (TNF-α) [28,29][28][29].
IFN-Is were extensively used to treat chronic inflammatory diseases, including autoimmune disorders such as MS, chronic viral infections, and malignant tumors. However, IFN-I-based treatments also produce significant adverse effects, including neurological and neuropsychiatric disorders and various systemic autoimmune diseases, such as rheumatoid arthritis, Aicardi–Goutières syndrome (AGS), systemic lupus erythematosus, Sjogren’s syndrome, and systemic sclerosis etc. [30]. Additionally, inhibition of B-cell activity or production of immunosuppressive molecules (e.g., IL-10) were reported by high concentrations of IFN-I treatment of long-term viral infections [14]. Therefore, IFN-I has pleiotropic effects, mobilizing immune cells to destroy viruses and bacteria on one hand and inducing neuroinflammation on the other. These complications or diseases have been termed “type I interferonopathies”. The following will discuss in more detail the beneficial and detrimental effects of type-I IFNs in the nervous system.
3. Role of IFN-Is in Long-Haul COVID Syndrome
Acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that started in Wuhan, China, at the end of 2019 has become a global pandemic, termed COVID-19. Both SARS-CoV-2 and SARS-CoV enter human host cells via the angiotensin-converting enzyme 2 (ACE2) receptor. Upon cell entry, sensing of coronaviruses by various pathogen recognition receptors, including TLRs and RLRs, can trigger innate immune responses via activation of the transcription factors nuclear factor-kB (NF-kB) and interferon regulatory factor 3 and 7 (IRF3, IRF7), which will stimulate the production of pro-inflammatory cytokines and IFN-I and type III IFNs (Figure 3). These interferons are translated and secreted from the infected cells in an autocrine or paracrine manner. Upon binding type I IFN to the IFNAR1/IFNAR2 receptor and type III IFN to the IFNLR1/IL-10R2 receptor, signal transduction is initiated, resulting in the formation of the ISGF3 complex (IRF9/p-STAT1/p-STAT2). This complex then induces ISG expression. Through a variety of mechanisms, ISGs suppress viral replication by preventing viral entry and viral trafficking into the cell nucleus, inhibiting transcription/translation and degrading viral nucleic acids and blocking viral particle assembly. Compared to type III IFNs, IFN-I signaling induces a stronger and faster ISG response and may confer protection against acute virus infection [102][31]. (Figure 3 and Figure 4).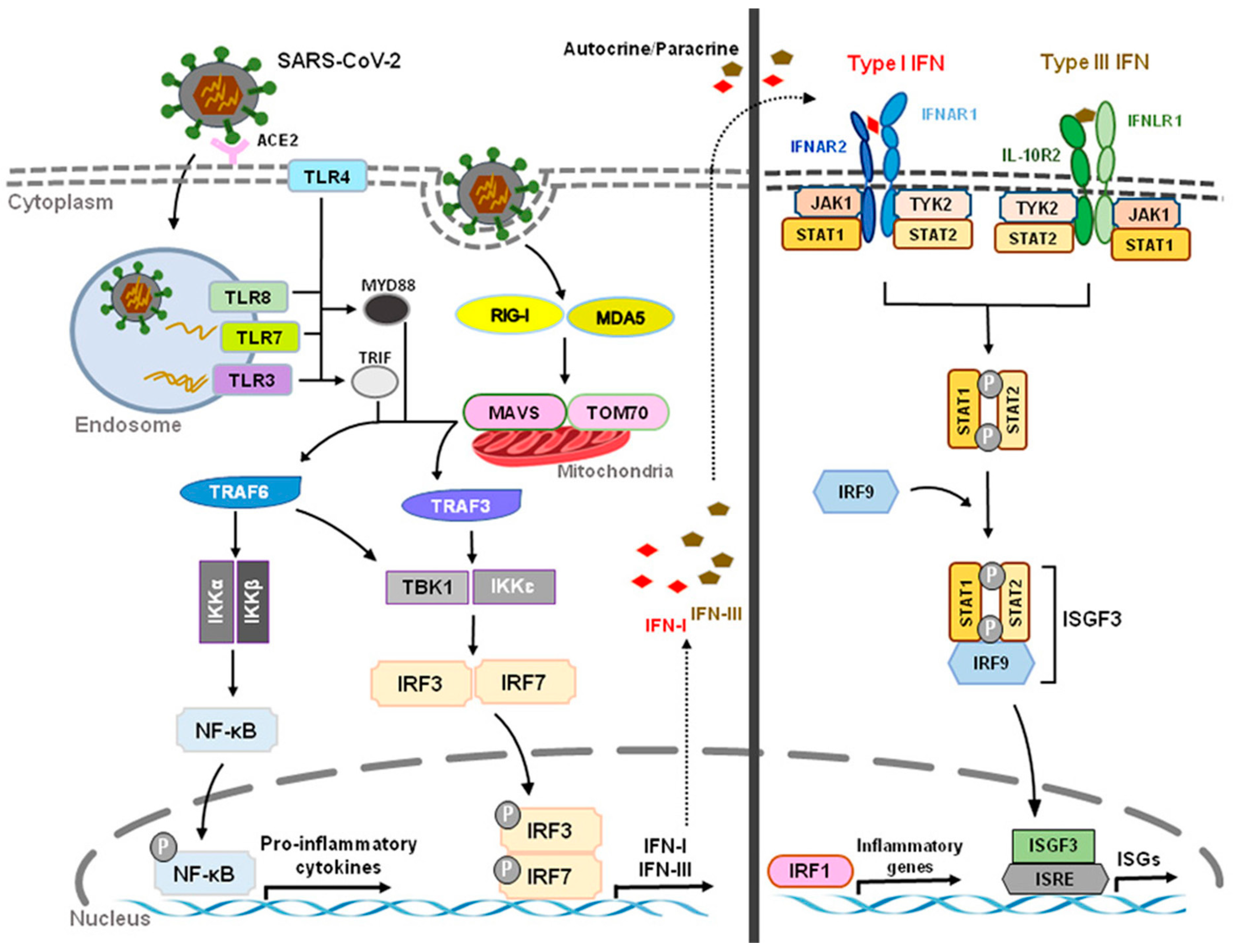
Figure 3. Innate Recognition and Interferon Signaling by Coronaviruses. Upon binding to angiotensin-converting enzyme 2 (ACE2) and subsequent sensing of coronaviruses by various pathogen recognition receptors, including toll-like receptors (TLRs) (TLR3, TLR4, TLR7, TLR8) and RIG-I-like receptors (RLRs) (RIG-I, MDA5), activation of transcription factors nuclear factor-kB (NF-kB) and interferon regulatory factor 3 and 7 (IRF3, IRF7) stimulates the production of pro-inflammatory cytokines and type I and III interferons (IFNs), respectively. The JAK/STAT signaling pathway is activated by IFNs through autocrine and paracrine secretion to induce the expression of IFN-stimulated genes (ISGs). Type I IFNs and IFN III IFNs are both capable of inducing ISGs, but type I IFN signaling produces a stronger and faster response as well as inducing pro-inflammatory cytokines and chemokines. However, delayed IFN-I responses not only fail to control SARS-CoV-2 virus but can also cause chronic inflammation and tissue damage.
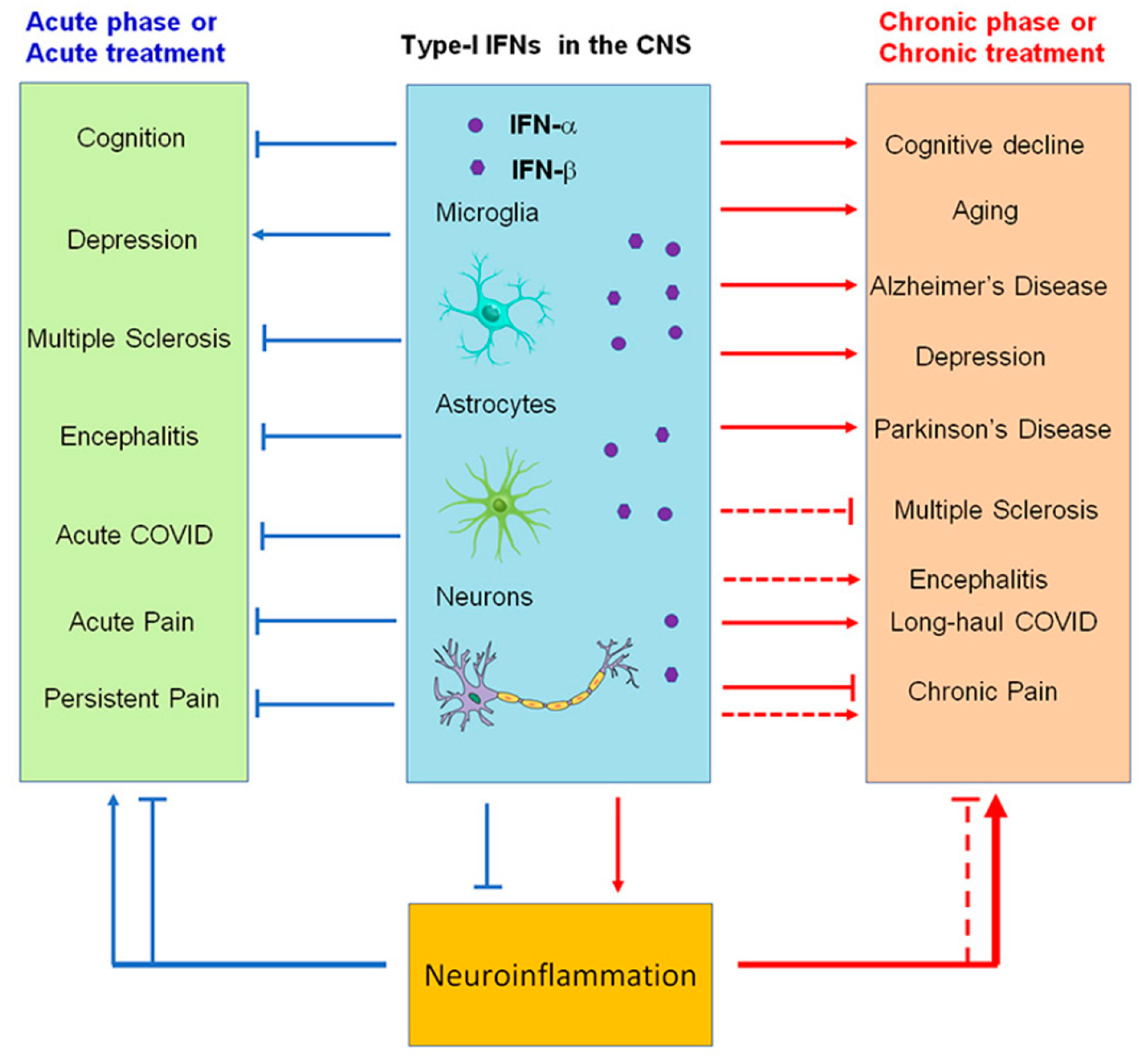
Figure 4. Modulation of neuroinflammation by type I IFNs in acute (blue) and chronic (red) neurological disease conditions. Note that IFN-α and IFN-β produce both beneficial and detrimental actions and may contribute to cognitive deficits (brain fog) in long-haul COVID.
References
- Pestka, S.; Langer, J.A.; Zoon, K.C.; Samuel, C.E. Interferons and their actions. Annu. Rev. Biochem. 1987, 56, 727–777.
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869.
- Pestka, S. The human interferon-alpha species and hybrid proteins. Semin. Oncol. 1997, 24, S9-4–S9-17.
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386.
- Donnelly, C.R.; Jiang, C.; Andriessen, A.S.; Wang, K.; Wang, Z.; Ding, H.; Zhao, J.; Luo, X.; Lee, M.S.; Lei, Y.L.; et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature 2021, 591, 275–280.
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Donnelly, C.R.; Chen, O.; Ji, R.R. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci. 2020, 43, 822–838.
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066.
- Liu, T.; Gao, Y.J.; Ji, R.R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144.
- Makris, S.; Paulsen, M.; Johansson, C. Type I Interferons as Regulators of Lung Inflammation. Front. Immunol. 2017, 8, 259.
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678.
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792.
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103.
- Asselin-Paturel, C.; Boonstra, A.; Dalod, M.; Durand, I.; Yessaad, N.; Dezutter-Dambuyant, C.; Vicari, A.; O’Garra, A.; Biron, C.; Brière, F.; et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2001, 2, 1144–1150.
- Biron, C.A. Interferons alpha and beta as immune regulators—A new look. Immunity 2001, 14, 661–664.
- Barchet, W.; Cella, M.; Odermatt, B.; Asselin-Paturel, C.; Colonna, M.; Kalinke, U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 2002, 195, 507–516.
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32.
- Taniguchi, T.; Takaoka, A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2001, 2, 378–386.
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23.
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445.
- Chen, J.; Baig, E.; Fish, E.N. Diversity and relatedness among the type I interferons. J. Interferon Cytokine Res. 2004, 24, 687–698.
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421.
- Levin, D.; Schneider, W.M.; Hoffmann, H.H.; Yarden, G.; Busetto, A.G.; Manor, O.; Sharma, N.; Rice, C.M.; Schreiber, G. Multifaceted activities of type I interferon are revealed by a receptor antagonist. Sci. Signal. 2014, 7, ra50.
- Dumitrescu, L.; Constantinescu, C.S.; Tanasescu, R. Recent developments in interferon-based therapies for multiple sclerosis. Expert Opin. Biol. Ther. 2018, 18, 665–680.
- Weise, A.M.; Flaherty, L.E. New options for the adjuvant treatment of cutaneous melanoma? Curr. Oncol. Rep. 2014, 16, 409.
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557.
- Benveniste, E.N.; Qin, H. Type I interferons as anti-inflammatory mediators. Sci. STKE 2007, 2007, pe70.
- Billiau, A. Anti-inflammatory properties of Type I interferons. Antivir. Res. 2006, 71, 108–116.
- Wang, H.; Wang, J.; Xia, Y. Defective Suppressor of Cytokine Signaling 1 Signaling Contributes to the Pathogenesis of Systemic Lupus Erythematosus. Front. Immunol. 2017, 8, 1292.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9.
- Peluso, M.J.; Kelly, J.D.; Lu, S.; Goldberg, S.A.; Davidson, M.C.; Mathur, S.; Durstenfeld, M.S.; Spinelli, M.A.; Hoh, R.; Tai, V.; et al. Rapid implementation of a cohort for the study of post-acute sequelae of SARS-CoV-2 infection/COVID-19. medRxiv 2021.
- Sudre, C.H.; Keshet, A.; Graham, M.S.; Joshi, A.D.; Shilo, S.; Rossman, H.; Murray, B.; Molteni, E.; Klaser, K.; Canas, L.S.; et al. Anosmia and other SARS-CoV-2 positive test-associated symptoms, across three national, digital surveillance platforms as the COVID-19 pandemic and response unfolded: An observation study. medRxiv 2020.
- Tenforde, M.W.; Billig Rose, E.; Lindsell, C.J.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Prekker, M.E.; Steingrub, J.S.; Smithline, H.A.; Gong, M.N.; et al. Characteristics of Adult Outpatients and Inpatients with COVID-19—11 Academic Medical Centers, United States, March-May 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 841–846.
- Darley, D.R.; Dore, G.J.; Byrne, A.L.; Plit, M.L.; Brew, B.J.; Kelleher, A.; Matthews, G.V. Limited recovery from post-acute sequelae of SARS-CoV-2 at 8 months in a prospective cohort. ERJ Open Res. 2021, 7, 384–2021.
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144.
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202.
- Remsik, J.; Wilcox, J.A.; Babady, N.E.; McMillen, T.A.; Vachha, B.A.; Halpern, N.A.; Dhawan, V.; Rosenblum, M.; Iacobuzio-Donahue, C.A.; Avila, E.K.; et al. Inflammatory Leptomeningeal Cytokines Mediate COVID-19 Neurologic Symptoms in Cancer Patients. Cancer Cell 2021, 39, 276–283.e3.
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216.
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B., Jr.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639.
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585.
- Chang, S.E.; Feng, A.; Meng, W.; Apostolidis, S.A.; Mack, E.; Artandi, M.; Barman, L.; Bennett, K.; Chakraborty, S.; Chang, I.; et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 5417.
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288.
- Zulfiqar, A.A.; Lorenzo-Villalba, N.; Hassler, P.; Andrès, E. Immune Thrombocytopenic Purpura in a Patient with COVID-19. N. Engl. J. Med. 2020, 382, e43.
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576.
- Qu, L.; Zhang, P.; LaMotte, R.H.; Ma, C. Neuronal Fc-gamma receptor I mediated excitatory effects of IgG immune complex on rat dorsal root ganglion neurons. Brain Behav. Immun. 2011, 25, 1399–1407.
- Qu, L.; Li, Y.; Pan, X.; Zhang, P.; LaMotte, R.H.; Ma, C. Transient receptor potential canonical 3 (TRPC3) is required for IgG immune complex-induced excitation of the rat dorsal root ganglion neurons. J. Neurosci. 2012, 32, 9554–9562.
- Wang, L.; Jiang, X.; Zheng, Q.; Jeon, S.M.; Chen, T.; Liu, Y.; Kulaga, H.; Reed, R.; Dong, X.; Caterina, M.J.; et al. Neuronal FcγRI mediates acute and chronic joint pain. J. Clin. Investig. 2019, 129, 3754–3769.
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A.; et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924.
- Ferini-Strambi, L.; Salsone, M. COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 2021, 268, 409–419.
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res. Ther. 2020, 12, 69.
- Taquet, M.; Luciano, S.; Geddes, J.R.; Harrison, P.J. Bidirectional associations between COVID-19 and psychiatric disorder: Retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry 2021, 8, 130–140.
- Naughton, S.X.; Raval, U.; Pasinetti, G.M. Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies. J. Alzheimer’s Dis. 2020, 76, 21–25.
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930.
- Bauer, K.; Schwarzkopf, L.; Graessel, E.; Holle, R. A claims data-based comparison of comorbidity in individuals with and without dementia. BMC Geriatr. 2014, 14, 10.
More