Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 4 by Jessie Wu.
Interstitial lung diseases (ILD) are relatively rare and sometimes become life threatening. In particular, rapidly progressive ILD, which frequently presents as acute lung injury (ALI) on lung histopathology, shows poor prognosis if proper and immediate treatments are not initiated. These devastating conditions include acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF), clinically amyopathic dermatomyositis (CADM), epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI)-induced lung injury, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infection named coronavirus disease 2019 (COVID-19).
- COVID-19
- ILD
- fibrosis
1. Introduction
Interstitial lung disease (ILD) is a relatively rare pathological condition that can induce respiratory insufficiency. In particular, rapidly progressive ILD frequently causes acute respiratory failure and death. Notably, diffuse alveolar damage (DAD) pattern on high-resolution computed tomography (HRCT) or on lung histopathological specimens are tied to a poor prognosis [1][2][3][4]. This rapidly progressive ILD with a poor prognosis includes acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF), clinically amyopathic dermatomyositis (CADM), epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI)-induced lung injury, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infection, named coronavirus disease 2019 (COVID-19). These devastating diseases frequently cause acute lung injury (ALI), including diffuse alveolar damage (DAD), although the damaged pulmonary cells, which sometimes become apoptotic, are quite different in each disease.
2. Clinically Amyopathic Dermatomyositis-Related Interstitial Lung Diseases
Connective tissue diseases (CTD) sometimes show lung involvement such as ILD, pulmonary hypertension, bronchiolitis, bronchiectasis/bronchiolectasis, and serositis, including pleuritis/pericarditis, depending on the CTD [5]. These lung involvements lead to a wide variety of lung opacities on high-resolution computed tomography (HRCT) and histopathological findings in lung specimens. As for prognosis, particularly the presence of ILD or pulmonary hypertension, is associated with poor survival [6][7][8]. However, the prognosis of patients with connective tissue diseases-interstitial lung diseases (CTD-ILD) is significantly better than that of IPF patients [9]. CTD-related ILD is frequently seen in patients with systemic sclerosis (SSc) or in those with polymyositis (PM)/dermatomyositis (DM) [5], and ILD accounts for 35% of the causes of death in SSc [6], 48% in polymyositis (PM)/ dermatomyositis (DM) [8], and 11% in rheumatoid arthritis (RA) [10]. PM/ dermatomyositis-interstitial lung disease (DM-ILD) progresses more rapidly [11][12] than systemic sclerosis-interstitial lung disease (SSc-ILD) [13] in many cases. Patients with PM/DM-ILD with acute/subacute onset show poorer survival than those with chronic onset [11][14][15]. In addition, DM-ILD shows worse survival than PM-ILD, and clinically amyopathic DM (CADM)-ILD has an even worse prognosis than DM-ILD or PM-ILD [12]. The definition of CADM includes both amyopathic and hypomyopathic DM [16]. Basic and clinical studies on disease-related antibodies have progressed in the field of myositis, and there are two major categories of autoantibodies: myositis-specific antibodies and myositis-associated antibodies [17]. The former includes anti-melanoma differentiation-associated gene (MDA) 5 antibody (previously called anti-CADM 140 antibody) and anti-amynoacyl-tRNA synthetase (ARS) antibodies. These two antibodies are exclusive and closely related to ILD rather than to myositis, although the prognoses differ widely among patients with each antibody. DM-ILD with anti-anti-amynoacyl-tRNA synthetase (ARS) antibody progresses slowly and responds well to corticosteroids; however, recurrence is not rare [15][18][19]. In contrast, DM-ILD with anti-MDA5 antibody progresses rapidly, is refractory to corticosteroid therapy, and shows poor survival [15][18][19][20][21]. MDA5 is a retinoic acid-inducible gene-I (RIG-I) family of intracellular viral sensors, and the positive rates of anti-MDA5 antibodies have been reported as 14.5–21.5% in DM [22][23] and 25% in PM/DM [15].
In patients with PM/DM and anti-MDA5 antibody, 53.3–82% of them have CADM [15][23][24][25], 94–95% have ILD [23][24], and 71–84% show rapidly progressive ILD [23] or acute/subacute (within 3 months) onset of ILD [15], which has a poor prognosis. Many patients with anti-MDA5 antibody live near the waterfront, and DM-ILD with anti-MDA5 antibody develops predominantly in October-March [25][26]. The exact reason for this is still unknown; however, MDA5 is known to recognise RNA viruses, and the induction of anti-MDA5 antibodies may be related to viral infection.
HRCT findings of DM-ILD with anti-MDA5 antibody are mainly consolidation/GGA and random GGA in lower lung field, and these opacities occupied 83.3% of all cases [27] (Figure 1A,B). Furthermore, 60% of these patients with such opacities on HRCT died despite intensive immunosuppressive treatments [27] (Figure 1C,D). Similarly, the HRCT findings of anti-MDA5 antibody-positive cases are mainly consistent with the unclassifiable pattern of IIPs (66.7%) [15]. As for lung histopathological pattern, five of six cases of DM or CADM had a DAD pattern, and all these patients with DAD died of the progression of ILD [3]. Similarly, seven of nine patients with CADM-ILD with anti-MDA5 antibody had DAD in the lung histopathological specimens [28].
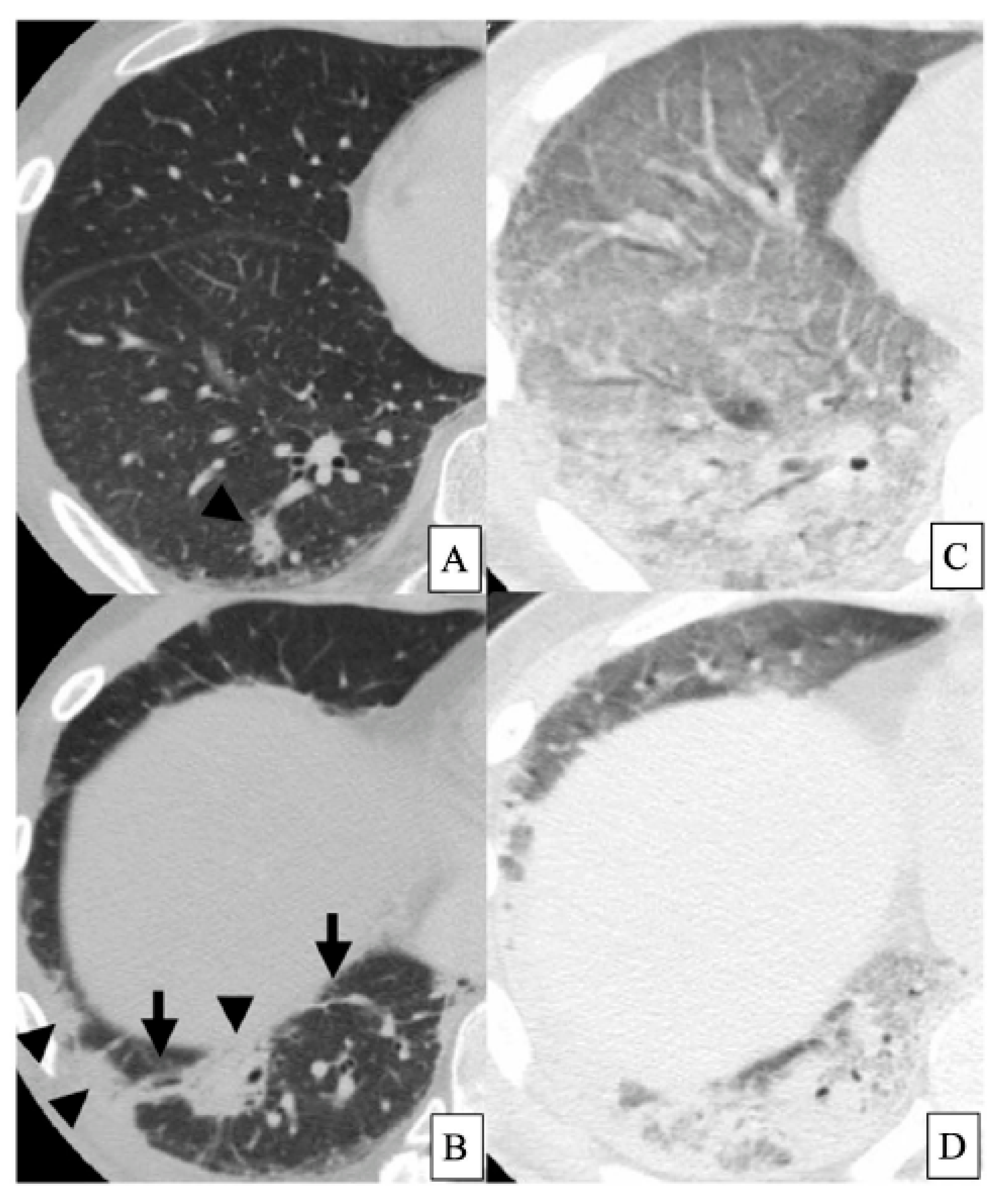
Figure 1. HRCT findings of the lungs in a patient with DM-ILD with anti-MDA5 antibody. A forty-four-year-old male patient with DM-ILD was positive for anti-MDA5 antibody. At diagnosis, peripheral and peribronchovascular consolidations are observed (arrowheads). Interlobular septal thickening and non-septal linear or plate-like opacities are also seen (arrows) ((A,B), from Tanizawa et al. [27] with permission). Despite treatments for 6 weeks, severe respiratory failure developed, requiring mechanical ventilation. Diffuse GGA and consolidation with air bronchograms are extended in the whole lungs (C,D) [27]. Surveillance at this point revealed no evidence of infection. The patient died of respiratory failure one week later. Abbreviations: HRCT, high-resolution computed tomography; DM, dermatomyositis; ILD, interstitial lung disease; MDA, anti-melanoma differentiation-associated gene; GGA, ground-glass attenuation.
Activated macrophages play a key role in the pathogenesis of CADM-ILD with anti-MDA5 antibody. Patients with CADM-ILD and anti-MDA5 antibody exhibit activated alveolar macrophages producing ferritin, similar to cells in the bone marrow, liver, and spleen [29]. These findings look similar to macrophage activation syndrome with hyperferritinemic syndrome [30]. In DM-ILD, increased serum ferritin levels negatively correlate with the P/F ratio [31], and patients with higher ferritin levels show a significantly worse survival and serum ferritin level is a significant prognostic factor [31][32]. In addition, in patients with DM-ILD and anti-MDA5 antibody, high macrophage hemophagocytic scores are related to higher ferritin levels [33]. The serum level of CD206, which is preferentially expressed on the surface of alternatively activated (M2) macrophages, was significantly increased in patients with CADM/DM-ILD and anti-MDA5 antibody [32]. M2 macrophages are closely related to tissue repair and fibrosis and the interaction between M2 macrophages and alveolar epithelial cells may be crucial [34]. Similarly, CD163 is also expressed on alveolar macrophages, especially on M2 macrophages [35], and serum levels of soluble CD163 are significantly higher in patients with DM-ILD than in those with PM or without ILD [36]. Furthermore, serum chitotriosidase [37], which macrophages/neutrophils produce, and YKL-40 [38], which is a chitinase family and macrophages/epithelial cells produce, increased in PM/DM-ILD, especially in DM-ILD with anti-MDA5 antibody. As for prognosis related with above mentioned molecules, higher level of serum ferritin [31][32], CD206 [32], CD163 [39], chitotriosidase [37], YKL-40 [38], and hemophagocytic scores [33] are tied to a poor prognosis. Taken together, as indicated above, activated macrophages contribute to the pathophysiology of DM-ILD with anti-MDA5 antibody. The role of monocytes in this process has also been investigated. CCL2 and interferon-induced protein with tetratricopeptide repeats (IFIT) 3 mRNA expression in monocytes and serum CCL2 and interferon (IFN)-β levels are increased in patients with DM-ILD and anti-MDA5 antibody [40]. In addition, serum CCL2 levels are significantly higher in patients with DM-ILD and anti-MDA5 antibody [41]. Both macrophages and monocytes produce CCL2, and the migration of monocytes to the lungs, depending on CCL2, may play a role in the pathogenesis of DM-ILD with anti-MDA5 antibody. In contrast, low circulating lymphocytes and monocytes are found in patients with DM-ILD and anti-MDA5 antibody [42]. In addition, higher serum CCL2 levels [41] and the lower numbers of lymphocytes and monocytes in peripheral blood [42] are associated with a poor survival. The H-ferritin subunit can inhibit lymphoid and myeloid cell proliferation via the T-cell immunoglobulin and mucin domains (TIM) 2 [35][43]. Therefore, H-ferritin produced by macrophages may be related to lower lymphocyte and monocyte counts in blood in patients with DM-ILD and anti-MDA5 antibody. Other immune cells and cytokines, such as neutrophils [44] and IL-8 [44][45], are also associated with a poor prognosis in patients with DM-ILD and anti-MDA5 antibody.
In terms of therapy, when PM/DM-ILD is slowly progressive with chronic onset without anti-MDA5 or anti-ARS antibodies, corticosteroids alone may be sufficient for treatment. However, when anti-MDA5 or anti-ARS antibodies are positive or patients show acute/subacute onset, corticosteroids and calcineurin inhibitors (tacrolimus or cyclosporine A) should be administered [46][47]. Furthermore, patients with acute/subacute onset and anti-MDA5 antibody or patients with rapidly progressive ILD should be treated with corticosteroids, calcineurin inhibitors, and intravenous cyclophosphamide (IVCY) [48][49]. This triple combination therapy significantly improves survival compared to historical controls [50]. However, triple combination therapy significantly increases the incidence of serious infections [50], with an odds ratio of 5.51 [51]. Therefore, overtreatment should be avoided in DM-ILD patients without rapidly progressive ILD or anti-MDA5 antibody [48][49]. Moreover, if DM-ILD is refractory to the above-mentioned treatments, intravenous immunoglobulin (IVIG) should be considered [52][53]. Finally, when DM or CADM-ILD progress despite appropriate treatment, this type of ILD is recently called progressive fibrosing (PF)-ILD [54] or progressive pulmonary fibrosis (PPF) [55]. If progressive cases meet the criteria for PF-ILD or PPF, patients with DM or CADM-ILD should be treated with an anti-fibrotic agent (nintedanib) [54] following the above-mentioned immunosuppressive treatments.
3. COVID-19 and Interstitial Lung Diseases
SARS-CoV2 is an enveloped positive-sense single-stranded RNA virus. The SARS-CoV2 outbreak occurred in Wuhan at the end of 2019 and spread worldwide thereafter. This infection by SARS-CoV2 was named COVID-19 and was declared to be a pandemic in March 2020 by the World Health Organization (WHO). As of October 2022, more than 613 million people have been infected with SARS-CoV2 and more than 6.5 million deaths have occurred worldwide (information on WHO COVID-19 dashboard: https://covid19.who.int). COVID-19 is now the most frequent infectious disease in the world far exceeding the prevalence of tuberculosis. The excess mortality rate exceeded 300 deaths per 100,000 people in 21 countries from January 2020 to December 2021 [56]. Notably, the mortality risk was higher in the Delta variant pandemic period than in the Omicron variant period [57]. Although the clinical presentation of COVID-19 is highly variable, some patients experience hypoxaemia without discomfort, which is called “silent hypoxemia” [58]. Thereafter, in some cases, COVID-19 causes severe acute respiratory distress syndrome (ARDS), particularly in patients with risk factors such as older age, male sex, cardiovascular disease, chronic respiratory disease, diabetes, obesity, and hypertension [59][60]. Patients with pre-existing ILD, such as IPF, RA-ILD, or SSc-ILD, show worse survival than those without pre-existing ILD [61]. Furthermore, patients with COVID-19-related AE-ILD have a worse prognosis than those without COVID-19 [62]. In patients without pre-existing ILD, those with COVID-19 ARDS have a higher body mass index and longer duration of mechanical ventilation than those with non-COVID-19 ARDS; however, 60-day mortality is similar [63]. In addition, thromboses such as pulmonary embolism and venous thromboembolism, which may be related to hypoxia, occur more frequently in COVID-19 especially in severe COVID-19 cases than in other infectious pneumonia or in mild to moderate COVID-19 cases, with an incidence of 9.5–30% [64][65][66]. Microthrombi in capillaries are found in fatal COVID-19-associated lung injuries [67]. On HRCT, multiple GGA, crazy paving patterns, and consolidation are seen in the peripheral or peribronchiolar lung area in many cases with COVID-19 [66][68] (Figure 2). These findings on HRCT change over time by phases (Figure 2A, early phase; B, 10 days later; and C, another 7 days later) [66], and the more diffuse extent of the findings on CT is associated with higher severity of COVID-19 [66]. Moreover, pulmonary vessel enlargement is observed in or adjacent to GGA and consolidation [66][69] (Figure 2D). Hypoxic pulmonary vasoconstriction (HPV) may be impaired due to endothelial damage caused by SARS-CoV-2 infection and may be related to pulmonary vessel enlargement on HRCT [60][70]. In addition, as mentioned above, thromboses such as pulmonary embolism are seen in severe COVID-19 patients (Figure 2E,F) [66]. In autopsy lung specimens of patients with fatal COVID-19, DAD patterns with excessive thrombosis and injury to alveolar epithelial cells/endothelial cells have been observed [60][71][72].
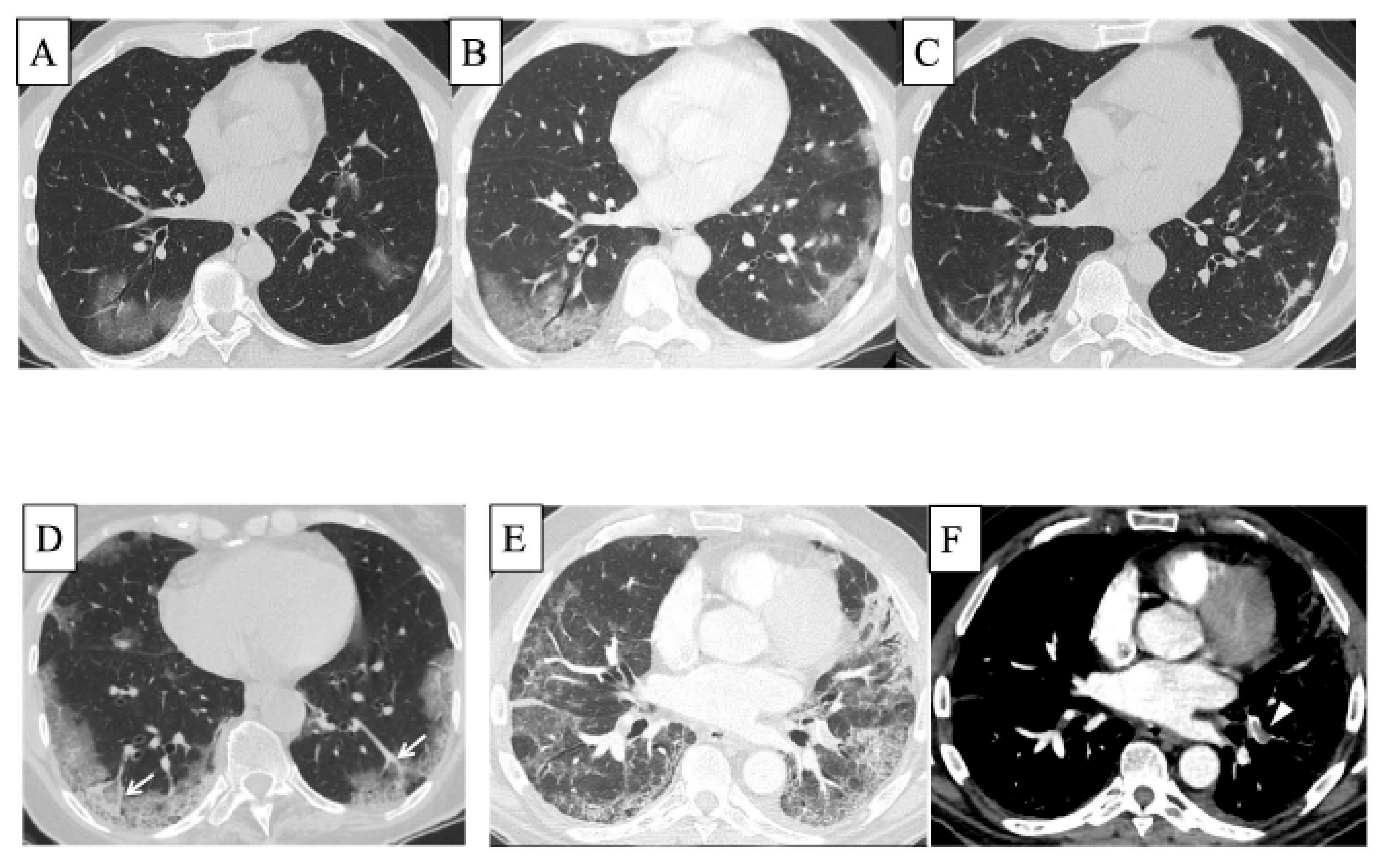
Figure 2. HRCT of the lungs in COVID-19. On HRCT, multiple GGA, crazy paving patterns, and consolidation are seen in peripheral or peribronchiolar lung area in many cases with COVID-19. These findings on HRCT change over time by phases, such as multiple GGA in the early phase (A), crazy paving appearance 10 days after the onset of symptoms in the progressive to peak phase (B), and multifocal consolidation with mild parenchymal distortion another 7 days later in absorption phase (C) (from Larici et al. [66] with permission). CT showing the presence of “enlarging vessel” sign within the areas of increased lung density (arrows, (D)) [66]. Acute pulmonary embolism is seen in severe COVID-19 patients (arrowhead, (E,F)) [66]. Abbreviations: HRCT, high-resolution computed tomography; COVID-19, coronavirus disease 2019; GGA, ground-glass attenuation.
SARS-CoV-2 invades human cells, including alveolar epithelial cells, vascular endothelial cells, and lymphocytes, via the angiotensin-converting enzyme (ACE) 2 receptor [73] and CD147 [74]. In the pathogenesis of COVID-19 and ILD. Cellular senescence and mitochondrial dysfunction play important roles [73]. Mitochondrial dysfunction and apoptosis are observed in lymphocytes, especially T cells, which are important for protection from SARS-CoV-2 [75], and are related to lymphocytopenia in patients with COVID-19 [74][76][77]. Mitochondrial ROS and subsequent activation of the NLRP3 inflammasome appear to be related to severe respiratory failure in coronavirus infection [78][79]. Furthermore, abnormal mitochondrial ultrastructure and increased expression of inhibitory checkpoints, such as programmed death-1 (PD-1) and its ligand (PD-L1), are found in monocytes in patients with COVID-19 [80]; and non-classical monocytes are further decreased in those with severe COVID-19 [81]. In line with these reports, T cells also show higher levels of the exhausted marker PD-1 and reduced expression of CXCR6 [82], which is important for the localisation of resident memory T cells, and the number of CD4+ and CD8+ T cells is reduced [83][84]. Moreover, the reduction in CD4+ and CD8+ T cells is negatively correlated with survival in patients with COVID-19 [84]. Additionally, cytokine production and reactivation of SARS-CoV-2 specific CD8+ T cells are inhibited in severe COVID-19 cases [85]. Similarly, apoptosis in plasmacytoid dendritic cells (DC) [86] and the decreased number of plasmacytoid DC and myeloid DC are found in patients with COVID-19 [87], which may be related to the impaired protective function by type 1 IFN. Additionally, neutrophils accumulate in the lungs, and calprotectin (S100A8/S100A9), which is a calcium-binding protein mainly produced from activated neutrophils, promotes inflammation [88] and increases in blood in patients with severe COVID-19 [81]. Neutrophils and neutrophil extracellular traps (NETs) are abundantly present in seriously damaged COVID-19 lung tissue [67], similar to ALI in influenza pneumonitis [89]. Regarding lung cell death, SARS-CoV-2 proteins and induced cytokines lead to PANoptosis consisting of apoptosis, pyroptosis, and necroptosis in the same cell population, which is a unique inflammatory programmed cell death [79][90]. SARS-CoV-2-induced synergistic effect of tumor necrosis factor (TNF)-α and IFN-γ causes PANoptosis [90]. Additionally, SARS-CoV-2-induced activation of caspase-8 causes apoptosis [91]. In this pathway, phosphorylation of receptor-interacting protein kinase-3 (RIPK3) and mixed lineage kinase domain-like (MLKL) also induces necroptosis [91] and facilitates inflammation via IL-1β, which may be related to COVID-19-induced ARDS. In fact, serum level of RIPK3 is significantly higher in severe COVID-19 cases than in mild cases [92]. Furthermore, the open reading frame (ORF) 3a, a SARS-CoV-2 accessory viroprotein, induces apoptosis via activation of caspase-8 [93] or enhances pyroptosis of infected cells [79].
In terms of prophylaxis, newly developed messenger RNA (mRNA) vaccines not only significantly reduce the number of infected patients [94][95], but also decrease the severity of COVID-19, although the efficacy rates to prevent infection decreased in the Omicron variant pandemic period in 2022 [57][96]. As for treatments, early use of antiviral medicines and late use of corticosteroids seem to be beneficial [97]. As of October 2022, in the viral replication phase, starting nirmatrelvir/ritonavir [97][98] or Molnupiravir [99] within 3–5 days of symptom onset reduce the risk of hospitalisation or death. Baricitinib [100], a Janus kinase (JAK) inhibitor, or Baricitinib with remdesivir [101] reduces the time to recovery or 28-day mortality, however, remdesivir alone does not reduce 28-day mortality [102]. Monoclonal antibodies target spike proteins and decrease symptom duration and mortality against the Delta variant but not against the Omicron variant, except for sotrovimab [97]. In the inflammatory phase, 6 mg/day of dexamethasone for 10 days decreases 28-day mortality in hospitalised patients with severe COVID-19 [103], although a higher dose of methylprednisolone may also be effective. Tocilizumab [104], an IL-6 receptor antagonist, and prophylactic anticoagulation [97] are also effective in critically ill patients with COVID-19.
References
- Oda, K.; Ishimoto, H.; Yamada, S.; Kushima, H.; Ishii, H.; Imanaga, T.; Harada, T.; Ishimatsu, Y.; Matsumoto, N.; Naito, K.; et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 109.
- Tanaka, K.; Enomoto, N.; Hozumi, H.; Isayama, T.; Naoi, H.; Aono, Y.; Katsumata, M.; Yasui, H.; Karayama, M.; Suzuki, Y.; et al. Serum S100A8 and S100A9 as prognostic biomarkers in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Investig. 2021, 59, 827–836.
- Matsuki, Y.; Yamashita, H.; Takahashi, Y.; Kano, T.; Shimizu, A.; Itoh, K.; Kaneko, H.; Mimori, A. Diffuse alveolar damage in patients with dermatomyositis: A six-case series. Mod. Rheumatol. 2012, 22, 243–248.
- Inoue, A.; Saijo, Y.; Maemondo, M.; Gomi, K.; Tokue, Y.; Kimura, Y.; Ebina, M.; Kikuchi, T.; Moriya, T.; Nukiwa, T. Severe acute interstitial pneumonia and gefitinib. Lancet 2003, 361, 137–139.
- Fischer, A.; du Bois, R. Interstitial lung disease in connective tissue disorders. Lancet 2012, 380, 689–698.
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airo, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Muller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815.
- Hoffmann-Vold, A.M.; Fretheim, H.; Halse, A.K.; Seip, M.; Bitter, H.; Wallenius, M.; Garen, T.; Salberg, A.; Brunborg, C.; Midtvedt, O.; et al. Tracking Impact of Interstitial Lung Disease in Systemic Sclerosis in a Complete Nationwide Cohort. Am. J. Respir. Crit. Care Med. 2019, 200, 1258–1266.
- Yamasaki, Y.; Yamada, H.; Ohkubo, M.; Yamasaki, M.; Azuma, K.; Ogawa, H.; Mizushima, M.; Ozaki, S. Longterm survival and associated risk factors in patients with adult-onset idiopathic inflammatory myopathies and amyopathic dermatomyositis: Experience in a single institute in Japan. J. Rheumatol. 2011, 38, 1636–1643.
- Park, J.H.; Kim, D.S.; Park, I.N.; Jang, S.J.; Kitaichi, M.; Nicholson, A.G.; Colby, T.V. Prognosis of fibrotic interstitial pneumonia: Idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med. 2007, 175, 705–711.
- Nakajima, A.; Inoue, E.; Tanaka, E.; Singh, G.; Sato, E.; Hoshi, D.; Shidara, K.; Hara, M.; Momohara, S.; Taniguchi, A.; et al. Mortality and cause of death in Japanese patients with rheumatoid arthritis based on a large observational cohort, IORRA. Scand. J. Rheumatol. 2010, 39, 360–367.
- Suda, T.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; Inui, N.; Naito, T.; Hashimoto, D.; Sato, J.; Toyoshima, M.; Hashizume, H.; et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur. Respir. J. 2006, 28, 1005–1012.
- Fujisawa, T.; Hozumi, H.; Kono, M.; Enomoto, N.; Hashimoto, D.; Nakamura, Y.; Inui, N.; Yokomura, K.; Koshimizu, N.; Toyoshima, M.; et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS ONE 2014, 9, e98824.
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528.
- Fujisawa, T.; Suda, T.; Nakamura, Y.; Enomoto, N.; Ide, K.; Toyoshima, M.; Uchiyama, H.; Tamura, R.; Ida, M.; Yagi, T.; et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J. Rheumatol. 2005, 32, 58–64.
- Hozumi, H.; Fujisawa, T.; Nakashima, R.; Johkoh, T.; Sumikawa, H.; Murakami, A.; Enomoto, N.; Inui, N.; Nakamura, Y.; Hosono, Y.; et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir. Med. 2016, 121, 91–99.
- Sontheimer, R.D. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis sine myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J. Am. Acad. Dermatol. 2002, 46, 626–636.
- Ghirardello, A.; Borella, E.; Beggio, M.; Franceschini, F.; Fredi, M.; Doria, A. Myositis autoantibodies and clinical phenotypes. Autoimmun. Highlights 2014, 5, 69–75.
- Hozumi, H.; Enomoto, N.; Kono, M.; Fujisawa, T.; Inui, N.; Nakamura, Y.; Sumikawa, H.; Johkoh, T.; Nakashima, R.; Imura, Y.; et al. Prognostic significance of anti-aminoacyl-tRNA synthetase antibodies in polymyositis/dermatomyositis-associated interstitial lung disease: A retrospective case control study. PLoS ONE 2015, 10, e0120313.
- Chen, F.; Li, S.; Wang, T.; Shi, J.; Wang, G. Clinical Heterogeneity of Interstitial Lung Disease in Polymyositis and Dermatomyositis Patients With or Without Specific Autoantibodies. Am. J. Med. Sci. 2018, 355, 48–53.
- Nakashima, R.; Imura, Y.; Kobayashi, S.; Yukawa, N.; Yoshifuji, H.; Nojima, T.; Kawabata, D.; Ohmura, K.; Usui, T.; Fujii, T.; et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology 2010, 49, 433–440.
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Aihara, K.; Ikezoe, K.; Watanabe, K.; Taguchi, Y.; Hatta, K.; et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir. Med. 2013, 107, 745–752.
- Labrador-Horrillo, M.; Martinez, M.A.; Selva-O’Callaghan, A.; Trallero-Araguas, E.; Balada, E.; Vilardell-Tarres, M.; Juarez, C. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J. Immunol. Res. 2014, 2014, 290797.
- Koga, T.; Fujikawa, K.; Horai, Y.; Okada, A.; Kawashiri, S.Y.; Iwamoto, N.; Suzuki, T.; Nakashima, Y.; Tamai, M.; Arima, K.; et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology 2012, 51, 1278–1284.
- Hoshino, K.; Muro, Y.; Sugiura, K.; Tomita, Y.; Nakashima, R.; Mimori, T. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology 2010, 49, 1726–1733.
- Nishina, N.; Sato, S.; Masui, K.; Gono, T.; Kuwana, M. Seasonal and residential clustering at disease onset of anti-MDA5-associated interstitial lung disease. RMD Open 2020, 6, e001202.
- Muro, Y.; Sugiura, K.; Hoshino, K.; Akiyama, M.; Tamakoshi, K. Epidemiologic study of clinically amyopathic dermatomyositis and anti-melanoma differentiation-associated gene 5 antibodies in central Japan. Arthritis Res. Ther. 2011, 13, R214.
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Watanabe, K.; Aihara, K.; Oga, T.; Chin, K.; Nagai, S.; et al. HRCT features of interstitial lung disease in dermatomyositis with anti-CADM-140 antibody. Respir. Med. 2011, 105, 1380–1387.
- Chino, H.; Sekine, A.; Baba, T.; Iwasawa, T.; Okudela, K.; Takemura, T.; Itoh, H.; Sato, S.; Suzuki, Y.; Ogura, T. Radiological and Pathological Correlation in Anti-MDA5 Antibody-positive Interstitial Lung Disease: Rapidly Progressive Perilobular Opacities and Diffuse Alveolar Damage. Intern. Med. 2016, 55, 2241–2246.
- Gono, T.; Miyake, K.; Kawaguchi, Y.; Kaneko, H.; Shinozaki, M.; Yamanaka, H. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology 2012, 51, 1336–1338.
- Rosario, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185.
- Gono, T.; Kawaguchi, Y.; Satoh, T.; Kuwana, M.; Katsumata, Y.; Takagi, K.; Masuda, I.; Tochimoto, A.; Baba, S.; Okamoto, Y.; et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology 2010, 49, 1713–1719.
- Horiike, Y.; Suzuki, Y.; Fujisawa, T.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; et al. Successful classification of macrophage-mannose receptor CD206 in severity of anti-MDA5 antibody positive dermatomyositis associated ILD. Rheumatology 2019, 58, 2143–2152.
- Zhao, S.; Ma, X.; Zhang, X.; Jin, Z.; Hu, W.; Hua, B.; Wang, H.; Feng, X.; Sun, L.; Chen, Z. Clinical significance of HScore and MS score comparison in the prognostic evaluation of anti-MDA5-positive patients with dermatomyositis and interstitial lung disease. Mod. Rheumatol. 2022, 32, 373–379.
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L709–L725.
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Astorri, E.; Perricone, C.; Blank, M.; Agmon-Levin, N.; Shoenfeld, Y.; Valesini, G. The hyperferritinemic syndromes and CD163: A marker of macrophage activation. Isr. Med. Assoc. J. 2014, 16, 662–663.
- Peng, Q.L.; Zhang, Y.L.; Shu, X.M.; Yang, H.B.; Zhang, L.; Chen, F.; Lu, X.; Wang, G.C. Elevated Serum Levels of Soluble CD163 in Polymyositis and Dermatomyositis: Associated with Macrophage Infiltration in Muscle Tissue. J. Rheumatol. 2015, 42, 979–987.
- Fujisawa, T.; Hozumi, H.; Yasui, H.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; Suda, T. Clinical Significance of Serum Chitotriosidase Level in Anti-MDA5 Antibody-positive Dermatomyositis-associated Interstitial Lung Disease. J. Rheumatol. 2019, 46, 935–942.
- Hozumi, H.; Fujisawa, T.; Enomoto, N.; Nakashima, R.; Enomoto, Y.; Suzuki, Y.; Kono, M.; Karayama, M.; Furuhashi, K.; Murakami, A.; et al. Clinical Utility of YKL-40 in Polymyositis/dermatomyositis-associated Interstitial Lung Disease. J. Rheumatol. 2017, 44, 1394–1401.
- Enomoto, Y.; Suzuki, Y.; Hozumi, H.; Mori, K.; Kono, M.; Karayama, M.; Furuhashi, K.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; et al. Clinical significance of soluble CD163 in polymyositis-related or dermatomyositis-related interstitial lung disease. Arthritis Res. Ther. 2017, 19, 9.
- Gono, T.; Okazaki, Y.; Kuwana, M. Antiviral proinflammatory phenotype of monocytes in anti-MDA5 antibody-associated interstitial lung disease. Rheumatology 2022, 61, 806–814.
- Oda, K.; Kotani, T.; Takeuchi, T.; Ishida, T.; Shoda, T.; Isoda, K.; Yoshida, S.; Nishimura, Y.; Makino, S. Chemokine profiles of interstitial pneumonia in patients with dermatomyositis: A case control study. Sci. Rep. 2017, 7, 1635.
- Lv, X.; Jin, Y.; Zhang, D.; Li, Y.; Fu, Y.; Wang, S.; Ye, Y.; Wu, W.; Ye, S.; Yan, B.; et al. Low Circulating Monocytes Is in Parallel with Lymphopenia Which Predicts Poor Outcome in Anti-melanoma Differentiation-Associated Gene 5 Antibody-Positive Dermatomyositis-Associated Interstitial Lung Disease. Front. Med. 2021, 8, 808875.
- Chen, T.T.; Li, L.; Chung, D.H.; Allen, C.D.; Torti, S.V.; Torti, F.M.; Cyster, J.G.; Chen, C.Y.; Brodsky, F.M.; Niemi, E.C.; et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med. 2005, 202, 955–965.
- Shirakashi, M.; Nakashima, R.; Tsuji, H.; Tanizawa, K.; Handa, T.; Hosono, Y.; Akizuki, S.; Murakami, K.; Hashimoto, M.; Yoshifuji, H.; et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology 2020, 59, 3284–3292.
- Gono, T.; Kaneko, H.; Kawaguchi, Y.; Hanaoka, M.; Kataoka, S.; Kuwana, M.; Takagi, K.; Ichida, H.; Katsumata, Y.; Ota, Y.; et al. Cytokine profiles in polymyositis and dermatomyositis complicated by rapidly progressive or chronic interstitial lung disease. Rheumatology 2014, 53, 2196–2203.
- Takada, K.; Katada, Y.; Ito, S.; Hayashi, T.; Kishi, J.; Itoh, K.; Yamashita, H.; Hirakata, M.; Kawahata, K.; Kawakami, A.; et al. Impact of adding tacrolimus to initial treatment of interstitial pneumonitis in polymyositis/dermatomyositis: A single-arm clinical trial. Rheumatology 2020, 59, 1084–1093.
- Fujisawa, T.; Hozumi, H.; Kamiya, Y.; Kaida, Y.; Akamatsu, T.; Kusagaya, H.; Satake, Y.; Mori, K.; Mikamo, M.; Matsuda, H.; et al. Prednisolone and tacrolimus versus prednisolone and cyclosporin A to treat polymyositis/dermatomyositis-associated ILD: A randomized, open-label trial. Respirology 2021, 26, 370–377.
- Fujisawa, T. Management of Myositis-Associated Interstitial Lung Disease. Medicina 2021, 57, 347.
- Kondoh, Y.; Makino, S.; Ogura, T.; Suda, T.; Tomioka, H.; Amano, H.; Anraku, M.; Enomoto, N.; Fujii, T.; Fujisawa, T.; et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir. Investig. 2021, 59, 709–740.
- Tsuji, H.; Nakashima, R.; Hosono, Y.; Imura, Y.; Yagita, M.; Yoshifuji, H.; Hirata, S.; Nojima, T.; Sugiyama, E.; Hatta, K.; et al. Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy with High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol. 2020, 72, 488–498.
- Sugiyama, Y.; Yoshimi, R.; Tamura, M.; Takeno, M.; Kunishita, Y.; Kishimoto, D.; Yoshioka, Y.; Kobayashi, K.; Takase-Minegishi, K.; Watanabe, T.; et al. The predictive prognostic factors for polymyositis/dermatomyositis-associated interstitial lung disease. Arthritis Res. Ther. 2018, 20, 7.
- Suzuki, Y.; Hayakawa, H.; Miwa, S.; Shirai, M.; Fujii, M.; Gemma, H.; Suda, T.; Chida, K. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung 2009, 187, 201–206.
- Wang, L.M.; Yang, Q.H.; Zhang, L.; Liu, S.Y.; Zhang, P.P.; Zhang, X.; Liu, X.J.; Han, L.S.; Li, T.F. Intravenous immunoglobulin for interstitial lung diseases of anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Rheumatology 2022, 61, 3704–3710.
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727.
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47.
- COVID-19 Excess Mortality Collaborators. Estimating excess mortality due to the COVID-19 pandemic: A systematic analysis of COVID-19-related mortality, 2020–2021. Lancet 2022, 399, 1513–1536.
- Adjei, S.; Hong, K.; Molinari, N.M.; Bull-Otterson, L.; Ajani, U.A.; Gundlapalli, A.V.; Harris, A.M.; Hsu, J.; Kadri, S.S.; Starnes, J.; et al. Mortality Risk Among Patients Hospitalized Primarily for COVID-19 during the Omicron and Delta Variant Pandemic Periods-United States, April 2020–June 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1182–1189.
- Simonson, T.S.; Baker, T.L.; Banzett, R.B.; Bishop, T.; Dempsey, J.A.; Feldman, J.L.; Guyenet, P.G.; Hodson, E.J.; Mitchell, G.S.; Moya, E.A.; et al. Silent hypoxaemia in COVID-19 patients. J. Physiol. 2021, 599, 1057–1065.
- Terada, M.; Ohtsu, H.; Saito, S.; Hayakawa, K.; Tsuzuki, S.; Asai, Y.; Matsunaga, N.; Kutsuna, S.; Sugiura, W.; Ohmagari, N. Risk factors for severity on admission and the disease progression during hospitalisation in a large cohort of patients with COVID-19 in Japan. BMJ Open 2021, 11, e047007.
- Swenson, K.E.; Swenson, E.R. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit. Care Clin. 2021, 37, 749–776.
- Zhao, J.; Metra, B.; George, G.; Roman, J.; Mallon, J.; Sundaram, B.; Li, M.; Summer, R. Mortality among Patients with COVID-19 and Different Interstitial Lung Disease Subtypes: A Multicenter Cohort Study. Ann. Am. Thorac. Soc. 2022, 19, 1435–1437.
- Kondoh, Y.; Kataoka, K.; Ando, M.; Awaya, Y.; Ichikado, K.; Kataoka, M.; Komase, Y.; Mineshita, M.; Ohno, Y.; Okamoto, H.; et al. COVID-19 and acute exacerbation of interstitial lung disease. Respir. Investig. 2021, 59, 675–678.
- Bain, W.; Yang, H.; Shah, F.A.; Suber, T.; Drohan, C.; Al-Yousif, N.; DeSensi, R.S.; Bensen, N.; Schaefer, C.; Rosborough, B.R.; et al. COVID-19 versus Non-COVID-19 Acute Respiratory Distress Syndrome: Comparison of Demographics, Physiologic Parameters, Inflammatory Biomarkers, and Clinical Outcomes. Ann. Am. Thorac. Soc. 2021, 18, 1202–1210.
- Nishimoto, Y.; Yachi, S.; Takeyama, M.; Tsujino, I.; Nakamura, J.; Yamamoto, N.; Nakata, H.; Ikeda, S.; Umetsu, M.; Aikawa, S.; et al. The current status of thrombosis and anticoagulation therapy in patients with COVID-19 in Japan: From the CLOT-COVID study. J. Cardiol. 2022, 80, 285–291.
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227.
- Larici, A.R.; Cicchetti, G.; Marano, R.; Bonomo, L.; Storto, M.L. COVID-19 pneumonia: Current evidence of chest imaging features, evolution and prognosis. Chin. J. Acad. Radiol. 2021, 4, 229–240.
- Obermayer, A.; Jakob, L.M.; Haslbauer, J.D.; Matter, M.S.; Tzankov, A.; Stoiber, W. Neutrophil Extracellular Traps in Fatal COVID-19-Associated Lung Injury. Dis. Markers 2021, 2021, 5566826.
- Iwasawa, T.; Sato, M.; Yamaya, T.; Sato, Y.; Uchida, Y.; Kitamura, H.; Hagiwara, E.; Komatsu, S.; Utsunomiya, D.; Ogura, T. Ultra-high-resolution computed tomography can demonstrate alveolar collapse in novel coronavirus (COVID-19) pneumonia. Jpn. J. Radiol. 2020, 38, 394–398.
- Lang, M.; Som, A.; Carey, D.; Reid, N.; Mendoza, D.P.; Flores, E.J.; Li, M.D.; Shepard, J.O.; Little, B.P. Pulmonary Vascular Manifestations of COVID-19 Pneumonia. Radiol. Cardiothorac. Imaging 2020, 2, e200277.
- Grimmer, B.; Kuebler, W.M. The endothelium in hypoxic pulmonary vasoconstriction. J. Appl. Physiol. 2017, 123, 1635–1646.
- D’Agnillo, F.; Walters, K.A.; Xiao, Y.; Sheng, Z.M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790.
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239.
- Torres Acosta, M.A.; Singer, B.D. Pathogenesis of COVID-19-induced ARDS: Implications for an ageing population. Eur. Respir. J. 2020, 56, 2002049.
- Helal, M.A.; Shouman, S.; Abdelwaly, A.; Elmehrath, A.O.; Essawy, M.; Sayed, S.M.; Saleh, A.H.; El-Badri, N. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J. Biomol. Struct. Dyn. 2022, 40, 1109–1119.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15.
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863.
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes. Infect. 2020, 9, 761–770.
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50.
- Paolini, A.; Borella, R.; De Biasi, S.; Neroni, A.; Mattioli, M.; Lo Tartaro, D.; Simonini, C.; Franceschini, L.; Cicco, G.; Piparo, A.M.; et al. Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells 2021, 10, 1585.
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001.
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18.
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534.
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635.
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827.
- Gangaev, A.; Ketelaars, S.L.C.; Isaeva, O.I.; Patiwael, S.; Dopler, A.; Hoefakker, K.; De Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; et al. Identification and characterization of a SARS-CoV-2 specific CD8(+) T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593.
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 2021, 184, 1836–1857.e22.
- Peruzzi, B.; Bencini, S.; Capone, M.; Mazzoni, A.; Maggi, L.; Salvati, L.; Vanni, A.; Orazzini, C.; Nozzoli, C.; Morettini, A.; et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS-CoV-2 infection. Immunology 2020, 161, 345–353.
- Ehrchen, J.M.; Sunderkotter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566.
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210.
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17.
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235.
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care 2020, 24, 484.
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615.
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416.
- Cohen, M.J.; Oster, Y.; Moses, A.E.; Spitzer, A.; Benenson, S.; Israeli-Hospitals 4th Vaccine Working Group. Association of Receiving a Fourth Dose of the BNT162b Vaccine with SARS-CoV-2 Infection among Health Care Workers in Israel. JAMA Netw Open 2022, 5, e2224657.
- Barthwal, M.S.; Dole, S.; Sahasrabudhe, T. Management of COVID-19: A comprehensive and practical approach. Med. J. Armed Forces India 2022.
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408.
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520.
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418.
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807.
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernandez Garcia, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for COVID-19-Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511.
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704.
- Investigators, R.-C.; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502.
More