Prolonged survival and durable responses in several late-stage cancers such as melanoma and lung cancer have been made possible with the use of immune checkpoint inhibitors targeting the programmed cell-death protein 1 (PD-1) or its ligand PD-L1. While it is prudent to focus on the unprecedented and durable clinical responses, there are subsets of cancer patients that do not respond to immunotherapies or respond early and then relapse later. Many pathways of resistance have been characterized, and more continue to be uncovered. To overcome the development of resistance, an in-depth investigation is necessary to identify alternative immune receptors and signals with the overarching goal of expanding treatment options for those with demonstrated resistance to PD1 checkpoint immunotherapy.
- cancer
- immunotherapy
- PD1
- PDL1
- resistance
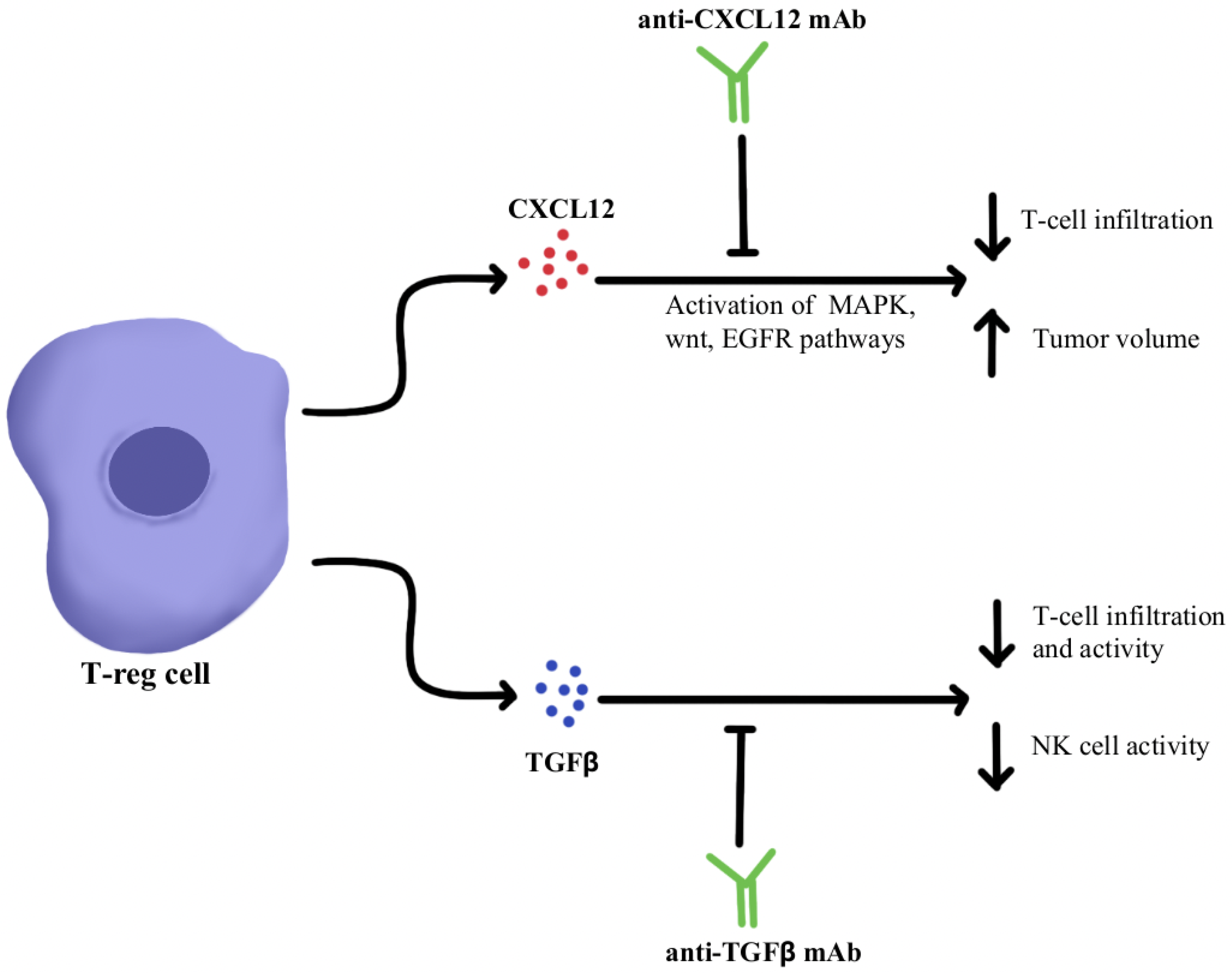
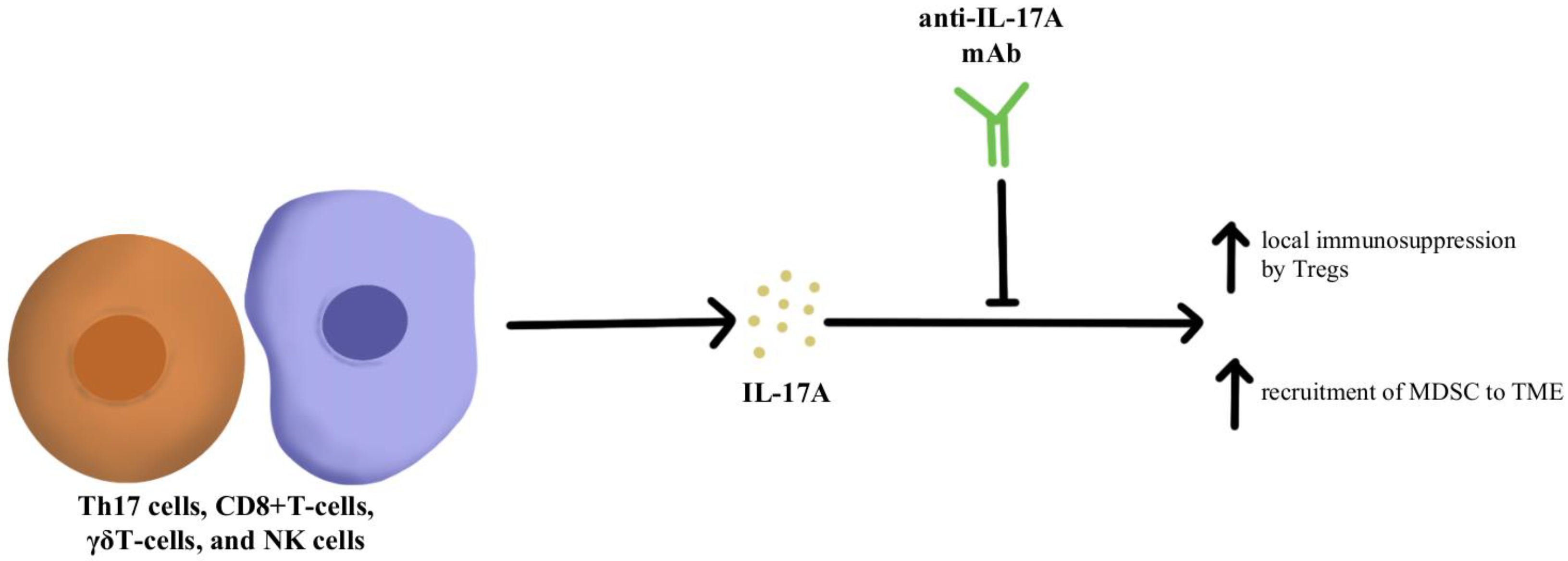
1. Immunosuppressive Cytokine Pathways
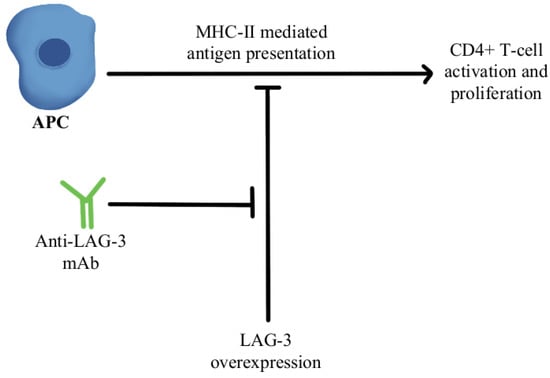
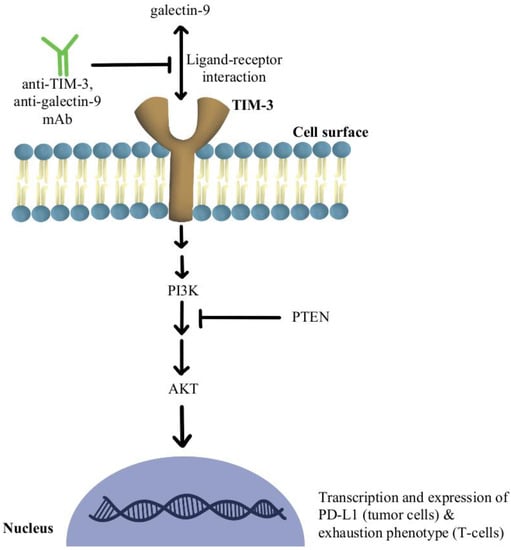
2. T-Cell Exhaustion and Depletion
3. Adoptive Cellular Therapy
4. Tumor Neoantigen Vaccines
References
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374.
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445.
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A Symbiotic Bridge Linking Cancer Cells and Their Stromal Neighbors in Oncogenic Communication Networks. Oncogene 2016, 35, 816–826.
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A Enhances Tumor Response to Anti-PD-1 Immunotherapy in Microsatellite Stable Colorectal Cancer. J. Immunother. Cancer 2021, 9, e001895.
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548.
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956.
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217.
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667.
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566.
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885.
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined Inhibition of CXCL12 and PD-1 in MSS Colorectal and Pancreatic Cancer: Modulation of the Microenvironment and Clinical Effects. J. Immunother. Cancer 2021, 9, e002505.
- Glasgow, E.; Mishra, L. Transforming Growth Factor-Beta Signaling and Ubiquitinators in Cancer. Endocr. Relat. Cancer 2008, 15, 59–72.
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of Signaling into CD4+ Cells via C3aR and C5aR Enables Autoinductive TGF-Β1 Signaling and Induction of Foxp3+ Regulatory T Cells. Nat. Immunol. 2013, 14, 162–171.
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual Targeting of TGF-β and PD-L1 via a Bifunctional Anti-PD-L1/TGF-ΒRII Agent: Status of Preclinical and Clinical Advances. J. Immunother. Cancer 2020, 8, e000433.
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.-Q.; Schlom, J.; Gameiro, S.R. M7824, a Novel Bifunctional Anti-PD-L1/TGFβ Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination with Vaccine. Oncoimmunology 2018, 7, e1426519.
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced Preclinical Antitumor Activity of M7824, a Bifunctional Fusion Protein Simultaneously Targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488.
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1287–1295.
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I Study of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Pretreated Biliary Tract Cancer. J. Immunother. Cancer 2020, 8, e000564.
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162.
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 2009, 31, 787–798.
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, Distribution, Generation, and Functional and Clinical Relevance of Th17 Cells in the Human Tumor Environments. Blood 2009, 114, 1141–1149.
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E. Interleukin-17 Inhibits Tumor Cell Growth by Means of a T-Cell-Dependent Mechanism. Blood 2002, 99, 2114–2121.
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. ΓδT17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800.
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 Inhibits CXCL9/10-Mediated Recruitment of CD8+ Cytotoxic T Cells and Regulatory T Cells to Colorectal Tumors. J. Immunother. Cancer 2019, 7, 324.
- Llosa, N.J.; Luber, B.; Tam, A.J.; Smith, K.N.; Siegel, N.; Awan, A.H.; Fan, H.; Oke, T.; Zhang, J.; Domingue, J.; et al. Intratumoral Adaptive Immunosuppression and Type 17 Immunity in Mismatch Repair Proficient Colorectal Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5250–5259.
- Brevi, A.; Cogrossi, L.L.; Grazia, G.; Masciovecchio, D.; Impellizzieri, D.; Lacanfora, L.; Grioni, M.; Bellone, M. Much More Than IL-17A: Cytokines of the IL-17 Family Between Microbiota and Cancer. Front. Immunol. 2020, 11, 565470.
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058.
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 Expression on HIV-Specific T Cells Is Associated with T-Cell Exhaustion and Disease Progression. Nature 2006, 443, 350–354.
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell 2014, 26, 923–937.
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684.
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927.
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954.
- Hosseinkhani, N.; Shadbad, M.A.; Asghari Jafarabadi, M.; Karim Ahangar, N.; Asadzadeh, Z.; Mohammadi, S.M.; Lotfinejad, P.; Alizadeh, N.; Brunetti, O.; Fasano, R.; et al. A Systematic Review and Meta-Analysis on the Significance of TIGIT in Solid Cancers: Dual TIGIT/PD-1 Blockade to Overcome Immune-Resistance in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 10389.
- Walker, L.S.K.; Sansom, D.M. The Emerging Role of CTLA4 as a Cell-Extrinsic Regulator of T Cell Responses. Nat. Rev. Immunol. 2011, 11, 852–863.
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory Mechanisms of Immune Checkpoints PD-L1 and CTLA-4 in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184.
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278.
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial Efficacy of Anti-Lymphocyte Activation Gene-3 (Anti–LAG-3; BMS-986016) in Combination with Nivolumab (Nivo) in Pts with Melanoma (MEL) Previously Treated with Anti–PD-1/PD-L1 Therapy. J. Clin. Oncol. 2017, 35, 9520.
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186.
- Huang, L.; Xu, Y.; Fang, J.; Liu, W.; Chen, J.; Liu, Z.; Xu, Q. Targeting STAT3 Abrogates Tim-3 Upregulation of Adaptive Resistance to PD-1 Blockade on Regulatory T Cells of Melanoma. Front. Immunol. 2021, 12, 654749.
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation Is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology 2017, 6, e1261779.
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216.
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3620–3629.
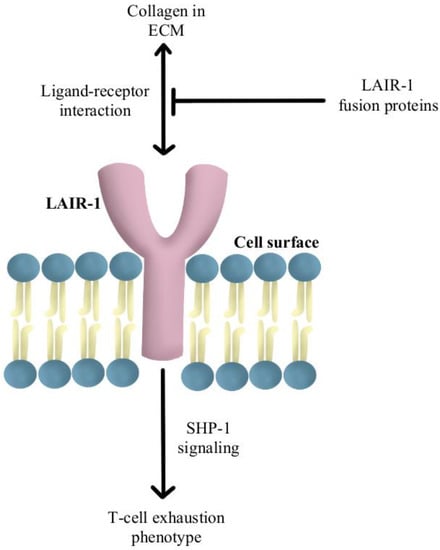
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen Promotes Anti-PD-1/PD-L1 Resistance in Cancer through LAIR1-Dependent CD8+ T Cell Exhaustion. Nat. Commun. 2020, 11, 4520.
- Lebbink, R.J.; van den Berg, M.C.W.; de Ruiter, T.; Raynal, N.; van Roon, J.A.G.; Lenting, P.J.; Jin, B.; Meyaard, L. The Soluble Leukocyte-Associated Ig-like Receptor (LAIR)-2 Antagonizes the Collagen/LAIR-1 Inhibitory Immune Interaction. J. Immunol. 2008, 180, 1662–1669.
- Meyaard, L.; Adema, G.J.; Chang, C.; Woollatt, E.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. LAIR-1, a Novel Inhibitory Receptor Expressed on Human Mononuclear Leukocytes. Immunity 1997, 7, 283–290.
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435.
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1136.
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829.
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14.
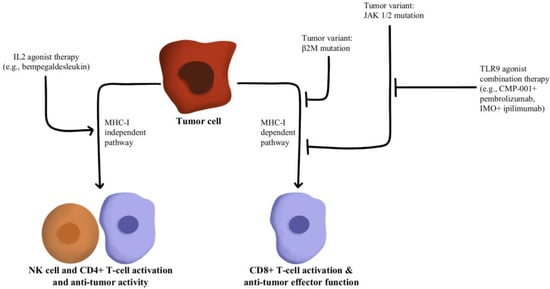
- Hurwitz, M.E.; Cho, D.C.; Balar, A.V.; Curti, B.D.; Siefker-Radtke, A.O.; Sznol, M.; Kluger, H.M.; Bernatchez, C.; Fanton, C.; Iacucci, E.; et al. Baseline Tumor-Immune Signatures Associated with Response to Bempegaldesleukin (NKTR-214) and Nivolumab. J. Clin. Oncol. 2019, 37, 2623.
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.-E.; Janssen, L.M.E.; Chesson, B.C.; Creasy, C.A.; Forget, M.-A.; Kahn, L.M.S.; Pazdrak, B.; et al. Bempegaldesleukin Selectively Depletes Intratumoral Tregs and Potentiates T Cell-Mediated Cancer Therapy. Nat. Commun. 2020, 11, 661.
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157.
- Diab, A.; Rahimian, S.; Haymaker, C.L.; Bernatchez, C.; Andtbacka, R.H.I.; James, M.; Johnson, D.B.; Markowitz, J.; Murthy, R.; Puzanov, I.; et al. A Phase 2 Study to Evaluate the Safety and Efficacy of Intratumoral (IT) Injection of the TLR9 Agonist IMO-2125 (IMO) in Combination with Ipilimumab (Ipi) in PD-1 Inhibitor Refractory Melanoma. J. Clin. Oncol. 2018, 36, 9515.
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive Biomarkers and Mechanisms Underlying Resistance to PD1/PD-L1 Blockade Cancer Immunotherapy. Mol. Cancer 2020, 19, 19.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12.
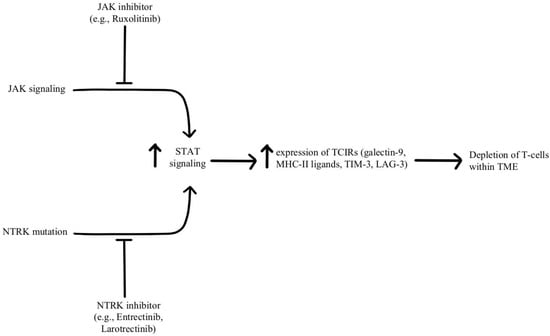
- Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/P53 Mutant Lung Cancer. Cancers 2019, 11, 462.
- US Food & Drug. Administration : FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 19 April 2022).
- Mian, A.; Hill, B.T. Brexucabtagene Autoleucel for the Treatment of Relapsed/Refractory Mantle Cell Lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441.
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients after Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56.
- Wang, J.; Deng, Q.; Jiang, Y.-Y.; Zhang, R.; Zhu, H.-B.; Meng, J.-X.; Li, Y.-M. CAR-T 19 Combined with Reduced-Dose PD-1 Blockade Therapy for Treatment of Refractory Follicular Lymphoma: A Case Report. Oncol. Lett. 2019, 18, 4415–4420.
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Front. Oncol. 2019, 9, 767.
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 Immunosuppression Boosts CAR T-Cell Therapy. Oncoimmunology 2013, 2, e26286.
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-Specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but Can Be Protected From Activation-Induced Cell Death by PD-1 Blockade. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1135–1149.
- Marotte, L.; Simon, S.; Vignard, V.; Dupre, E.; Gantier, M.; Cruard, J.; Alberge, J.-B.; Hussong, M.; Deleine, C.; Heslan, J.-M.; et al. Increased Antitumor Efficacy of PD-1-Deficient Melanoma-Specific Human Lymphocytes. J. Immunother. Cancer 2020, 8, e000311.
- Marotte, L.; Capitao, M.; Deleine, C.; Beauvais, T.; Cadiou, G.; Perrin, J.; Chérel, M.; Scotet, E.; Guilloux, Y.; Bruchertseifer, F.; et al. Anti-Tumor Efficacy of a Combination Therapy with PD-L1 Targeted Alpha Therapy and Adoptive Cell Transfer of PD-1 Deficient Melanoma-Specific Human T-Lymphocytes. Oncoimmunology 2021, 10, 1940676.
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-Tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118.
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737.
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. CII 2019, 68, 365–377.
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6982–6992.
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved Survival of Chimeric Antigen Receptor-Engineered T (CAR-T) and Tumor-Specific T Cells Caused by Anti-Programmed Cell Death Protein 1 Single-Chain Variable Fragment-Producing CAR-T Cells. Cancer Sci. 2019, 110, 3079–3088.
- Suarez, E.R.; Chang, D.K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget 2016, 7, 34341–34355.
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat. Rev. 2017, 52, 71–81.
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. The Oncologist 2015, 20, 812–822.
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen Vaccine: An Emerging Tumor Immunotherapy. Mol. Cancer 2019, 18, 128.
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.-C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma-A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832.
- Nesselhut, J.; Marx, D.; Lange, H.; Regalo, G.; Cillien, N.; Chang, R.Y.; Nesselhut, T. Systemic Treatment with Anti-PD-1 Antibody Nivolumab in Combination with Vaccine Therapy in Advanced Pancreatic Cancer. J. Clin. Oncol. 2016, 34, 3092.
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. M6A Regulator-Mediated Methylation Modification Patterns and Tumor Microenvironment Infiltration Characterization in Gastric Cancer. Mol. Cancer 2020, 19, 53.
- Nemunaitis, J. Vaccines in Cancer: GVAX, a GM-CSF Gene Vaccine. Expert Rev. Vaccines 2005, 4, 259–274.
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973.
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013, 73, 3591–3603.
- Chen, C.-Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic Virus and PD-1/PD-L1 Blockade Combination Therapy. Oncolytic Virotherapy 2018, 7, 65–77.