Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Martina De Angelis and Version 5 by Jessie Wu.
Hydroarylation reactions enable the formation of new Csp2–Csp3 or Csp2–Csp2 bonds using aromatic substrates. Its outcome is described as the addition of hydrogen and an aryl group to an unsaturated moiety, resulting in the functionalization of the aromatic Csp2–H bond. Hydroarylation reactions can occur by a direct functionalization via insertion of an unsaturated compound or with the use of pre-activated aryl substrates, usually aryl-iodides or aryl-boronated. Below are reported some example from the scientific production of the last 10 years about the asymmetric hydroarylation of activated aryl portions. N In this context, nickel and palladium turned out to be the more-employed metals.
- nickel
- asymmetric hydroarylation
- palladium
1. Palladium
The palladium-catalyzed hydroarylation between olefines and arylhalides or pseudohalides was reported for the first time by Cacchi and co-workers and is formally known as a reductive Heck reaction [1][2][3][4][51,52,53,54]. Although its disclosure dates back to the 1980s, its asymmetric version has only been developed recently. Between 2010 and 2011 Sigman described the hydroarylation of dienes and styrenes trough the reductive formation of π-allyl palladium intermediates under oxidative conditions [5][6][55,56]. In 2013 Liu and Zhou reported the desymmetrization of substituted cyclopentenes through an asymmetric Heck reaction and the first highly enantioselective hydroarylation of bicyclic olefins (1a and 1b) with a Pd catalyst [7][57] (Figure 1). For the first time in the asymmetric hydroarylation catalyzed by Pd, the reaction was achieved with high yields and ee up to 90% in all cases.
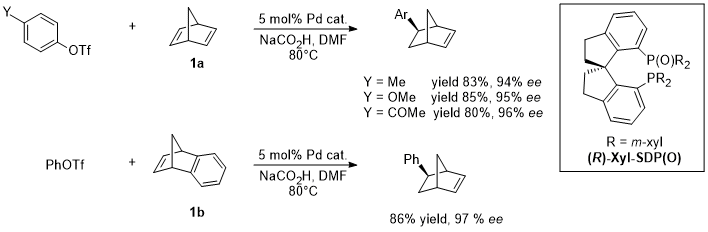
Figure 1.
Asymmetric hydroarylation catalyzed by Pd/(R)-Xyl-SDP(O).
The combination of Cu and Pd as cooperative catalysis in the asymmetric hydroarylation or hydrocyanation is described by the works of Buchwald’s group [8][9][10][58,59,60]. The asymmetric hydroarylation of 2 and 5 (Figure 2) leads to the synthesis of 1,1-diarylalkanes (4) and of arenes with β-stereogenic center (7) . In Figure 3, the complete catalytic cycle is reported: simultaneously with the copper activity, the Pd(0) complex is oxidatively added to the aryl bromide (8), forming the aryl palladium intermediate (9), which converges in a stereospecific transmetalation with the organocopper species (12). The enantioenriched products (11) are finally furnished via reductive elimination of chiral Pd(II) alkyl complexes (10), regenerating the Pd(0)-Ligand compound. After testing several ligands for both the processes, the BrettPhos for the Pd and the DTBM-SEGPHOS for the Cu turned out to be the best ligands in terms of yields and enantioselectivity.
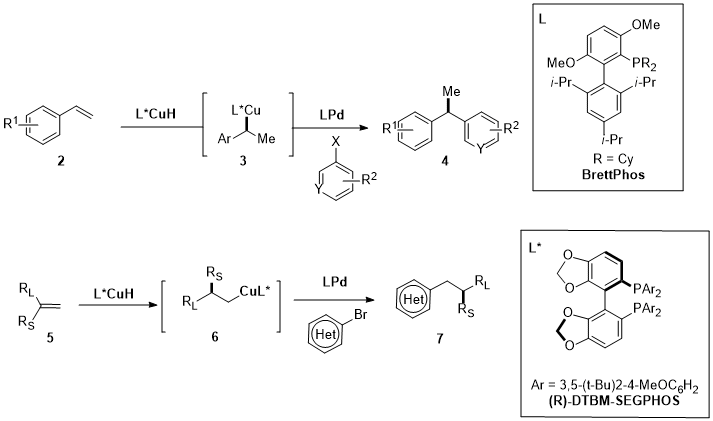
Figure 2. Asymmetric hydroarylation by Cu/Pd cooperative catalysis for the synthesis of 1,1-diarylalkanes (4) (a) and for the synthesis of arenes with β-stereogenic center (7) (b).
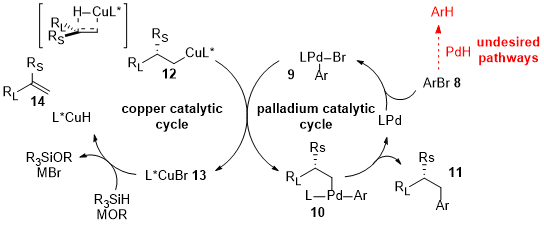
Figure 3.
Proposed catalytic cycle of reaction in
Figure 2
b.
In a recent work, the issue of the employment of two different ligands in the asymmetric hydroarylation through the dual Pd-CuH catalysis was overcome using the DTBM-SEGPHOS for both the metals. The reaction was performed on numerous 1,1-disubstituted alkenes and aryl bromides providing access in a single-step process to arenes with β-stereogenic centers, motifs founded in many drugs and natural products [11][12][13]. Zhu and co-workers developed an asymmetric intramolecular reductive Heck reaction of the issue of the employment of two different ligands in the asymmetric hydroarylation through the dual Pd-CuH catalysis was overcome using the DTBM-SEGPHOS for both the metals. The reaction was performed on numerous 1,1-disubstituted alkenes and aryl bromides providing access in a single-step process to arenes with β-stereogenic centers, motifs founded in many drugs and natural products [61,62,63]. Zhu and co-workers developed an asymmetric intramolecular reductive Heck reaction of N-aryl acrylamides [14]. Synthesis of enantioenriched 3,3-disubstituted oxindoles (-aryl acrylamides [64]. Synthesis of enantioenriched 3,3-disubstituted oxindoles (16) with a quaternary stereocenter was achieved with high yields and ee. Choice of the chiral ligand determines not only the enantioselectivity of the reaction, but also the reaction pathways: when PPh3 is used the reaction leads to the carboborylation product, while using (S)-tBuPHOX as ligand, the reaction pathway switches, affording hydroarylation products (Figure 4).
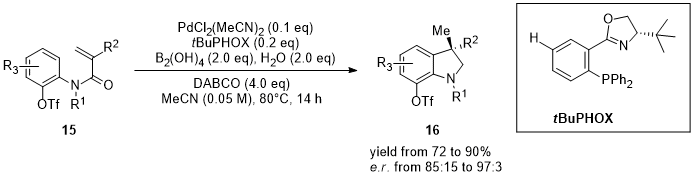
Figure 4.
Asymmetric intramolecular reductive Heck reaction of
N
-aryl acrylamides (
15
).
Zhang’s group reported the first example of highly enantioselective intramolecular hydroarylation of allyl aryl ethers through a reductive Heck reaction catalyzed by palladium [15][65]. The reaction was performed exploiting a new chiral sulfonamide monophosphine ligand, N-Me-XuPhos. The hydroarylation of allyl aryl ethers (17) using N-Me-XuPhos ligands, afforded a variety of optically active 3,3-Disubstituted 2,3-Dihydrobenzofuran (18) (Figure 5) and a series of CB2 receptor agonists with high chemo- and enantioselectivity (70–91% yields with 91–95% ee) (Figure 6).
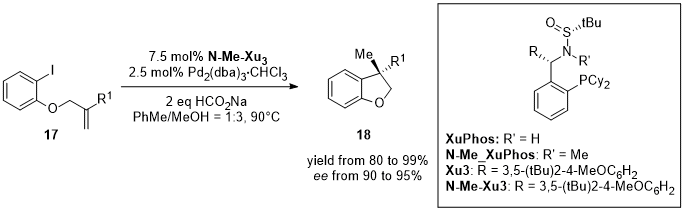
Figure 5.
Asymmetric intramolecular reductive Heck reaction of allyl aryl ethers (
17
).
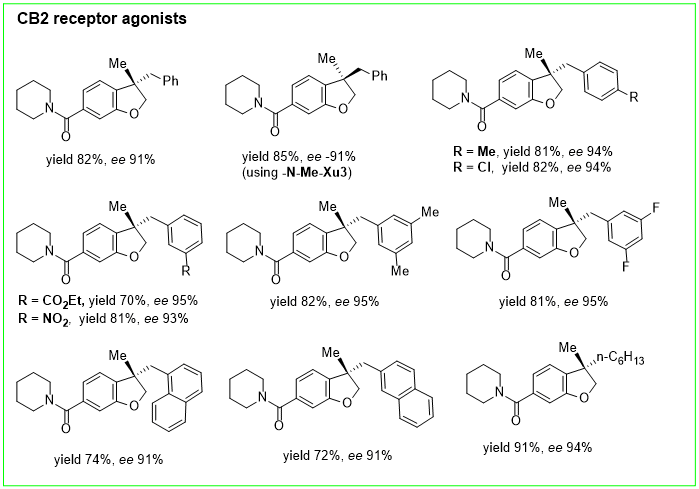
Figure 6.
CB2 receptor agonists obtained with synthesis in
Figure 5
.
Engle and coworkers have developed an enantioselective reductive Heck hydroarylation of alkenes that takes advantage of a chiral transient directing group (TDG) [16][66]. The chiral TDG class selected are chiral amines, in particular amino acids (L-tert-leucine in Figure 7), which lead to a stereocontrolled migratory insertion with alkenyl benzaldehydes (19) under mild conditions.
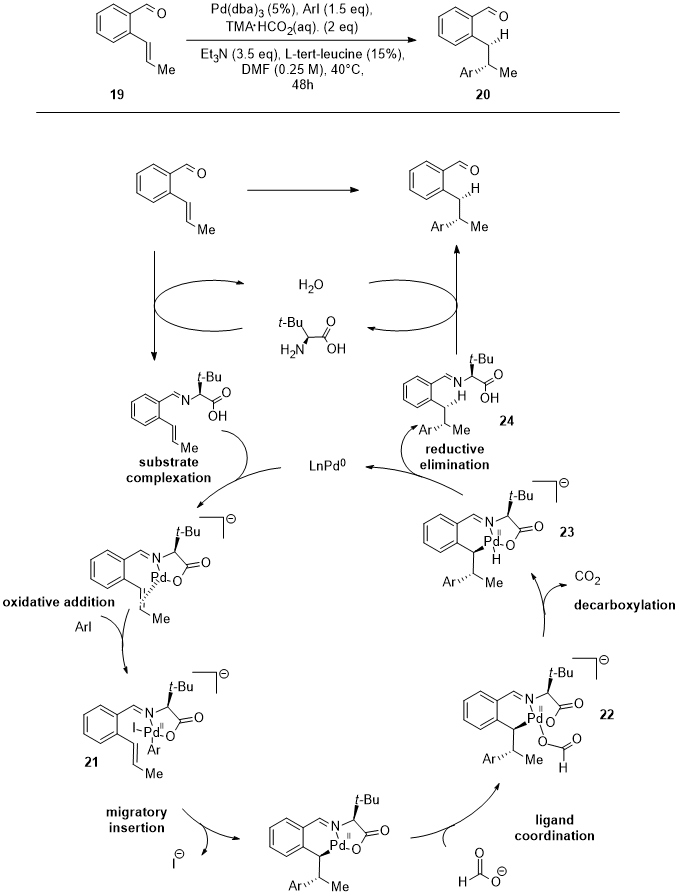
Figure 7.
Palladium reductive Heck hydroarylation of alkenes (
19
) assisted by L-tert-leucine.
2. Nickel
Hydroarylation reactions involving catalytic organoligand/Ni systems and pre-activated arene are more represented compared to the direct counterpart (Section 2.4). Between 2019 and 2021 Zhu’s group published several works in high-impact journals where the catalytic systems involved Nickel and preactivated arenes in hydroarylation reactions [17][18][19][20][21][67,68,69,70,71]. In this reaction, NiH species generated in situ catalyze both the chain walking process and subsequent cross-coupling, leading to α-functionalized alkyl boronate species with excellent chemo- and regioselectivity. Moreover, the asymmetric version of the reaction performed on these substrates by using (S,S)-Cy-Biox as the chiral ligand (L* in the scheme of Figure 8) delivered the product (26) in good yield but with low enantioselectivity.
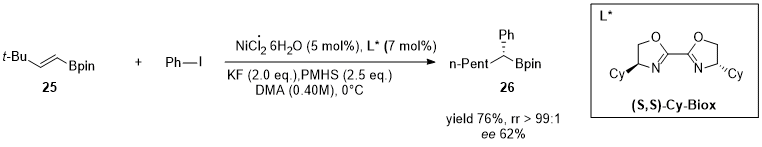
Figure 8.
Hydroarylation of boron containing alkene (
25
) and benzyl iodide by using Ni/(S,S)-Cy-Biox.
A highly enantio- and regioselective NiH-catalyzed reductive hydroarylation of vinylarenes (27) with aryl iodides was reported. The developed protocol involves the employment of a bis imidazoline (BIm)–nickel complex as catalyst. The proposed mechanism (Figure 9) shows that after formation of an alkyl–nickel intermediate (28) by the reaction between the catalyst and the alkene, the oxidative addition of aryl iodide occurs. The enantio-determining step of the process is due to the enantioselective recombination of the benzylic radical (31) with the Ar-Ni(II)-I complex (30). An irreversible reductive elimination of the Ar-Ni(III)–alkyl complex (29) provides an enantioenriched 1,1-diarylalkane product (32) and nickel(I) iodide (33).
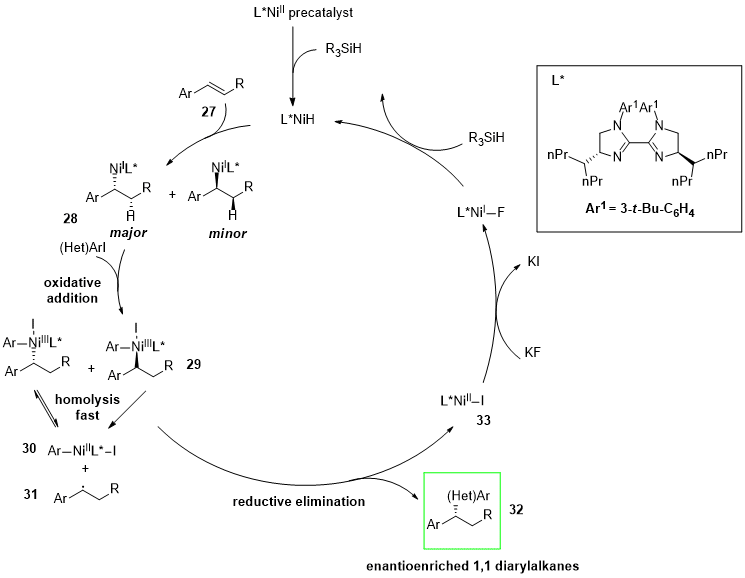
Figure 9.
NiH-catalyzed asymmetric reductive hydroarylation of vinylarenes (
27
) with aryl iodides.
The employment of chiral BIm-Ni systems in the asymmetric reductive hydroarylation of vinyl amides (34) for the synthesis of enantioenriched α-arylbenzamides (35) was reported by Nevado [22][72]. Different aromatic substituents on the ligand’s nitrogen atoms and isopropyl side chain were tested, leading to the product in moderate yields but with good enantioselectivity. The employment of the ligand L*IV and the optimization of the reaction conditions allowed for the obtaining of a wide variety of α-arylbenzamides in good yields and with high enantioselectivity (Figure 10).
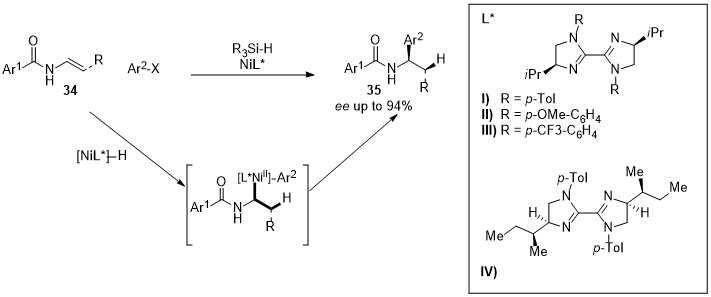
Figure 10.
Bis-imidazoline-Nickel complex employed to synthesize enantioenriched α-arylbenzamides (
35
).
The use of aryl- or vinylboronic species in the asymmetric hydroarylation/hydroalkenylation of styrenes and 1,3-dienes was reported by the research groups of Mei, Murcum and Zhou [23][24][25][73,74,75]. Stanley’s group recently reported the catalytic enantioselective hydroarylations of vinylarenes (36) with aryl boronic acids in the presence of a catalyst generated in situ aimed at the synthesis of chiral 1,1-diarylethanes (38) (Figure 11a) [26][76]. As reported in Figure 11b, the reactions performed with arylboronic acids having electron-withdrawing substitutents allowed the preparation of the corresponding 1,1-diarylethanes with less than 80% ee, while boronic acids having electron-donating groups gave the desired products with greater than 90% ee. 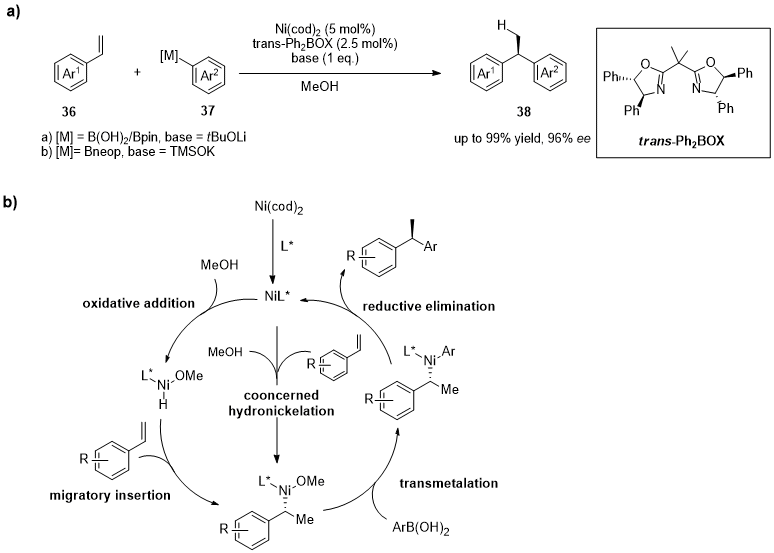
Figure 11. (a) Synthesis of chiral 1,1-diarylethanes (38) trough Ni/ trans-Ph2BOX. Catalyst are generated in situ. (b) Mechanism of concerted hydronickelation.
3. Rhodium
Asymmetric arylation reaction between aryl– or alkenyl–boronic acids and olefins has been widely studied by Hayashi and co-workers since the 90s [27][28][29][30][77,78,79,80]. Recently, Hayashi’s group has published a work about the Rh-catalyzed asymmetric hydroarylation of 3-pyrrolines [31][27]. The desired hydroarylated product was achieved with high chemoselectity and excellent enantiomeric excess. The asymmetric hydroarylation of 3-pyrrodines with arylboroxines provides a variety of 3-arylpyrrolidines which are important biologically active compounds.
Aryloboroxines ((ArBHO)3) (40) have been also used as key reagents in the asymmetric hydroarylation of divinylphosphine oxides (39) (RP(O)(CH=CH2)2) [32][81]. The reaction provides the corresponding monoarylated products (41 and 42) giving the deasymmetrization and the enantioselective formation of the product with a P-stereogenic center. (R)-DTBM-segphos was found to be the best ligand regarding chemo- and enantioselectivity (Figure 12). When the reaction is performed between the aryl boroxine and the divinylphosphine oxide with bulky substituents (R- in the list of Figure 12), high yields and ee in the formation of the product (R)-41 were observed, while the use of less bulky substituents leads to a lowering of yield and/or enantioselectivity. Concerning the substituted arylboroxines, para- or meta- substituents lead to product 41 in high yields with good enantioselectivity. Ortho-substitution leads to an increase in ee but also to a decrease in 41/42 selectivity, up to an inversion in favor of the product 42 (ratio 41/42 = 19/81) when the 2-fluorophenylboronic acid is used.
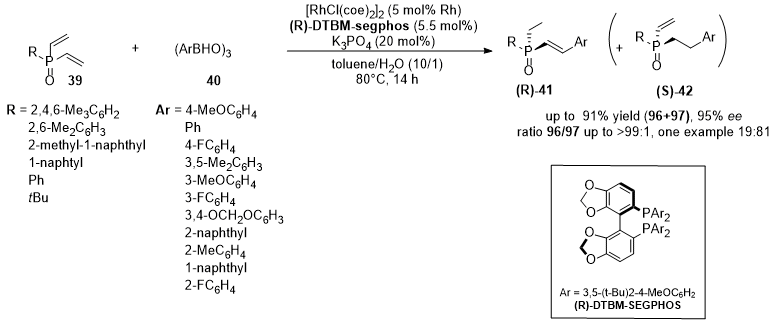
Figure 12.
Asymmetric hydroarylation of divinylphosphine oxides (
39
) and aryl boroxines (
40
). Catalyst complex: Rh/(R)-DTBM-segphos.
Wang and co-workers reported an example of Rh-catalyzed asymmetric hydroarylation for kinetic resolution and dynamic kinetic resolution of chromenes [33][82]. This strategy exploits the preferential coordination of the Rh-Ligand complex with one of the two enantiomers of the racemic mixture while the formation of the other intermediate is much less favorable because of steric repulsion (Figure 13). Therefore, one enantiomer (43) undergoes the transformation in 2,3-diaryl chromene (44) through the asymmetric hydroarylation reaction leaving the other optically pure unreacted flavene.
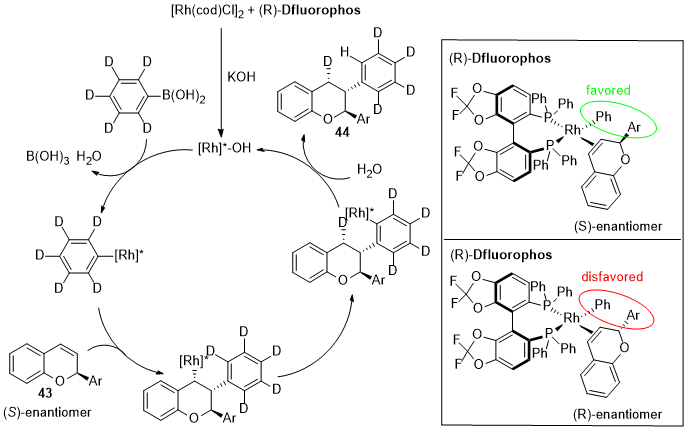
Figure 13.
Rhodium-catalyzed hydroarylation of 2-aryl chromenes: Deuterium-labeling experiment and proposed mechanism.
The screening of several biphosphines ligands showed that (R)-Difluorphos gave the best results. Good yields and excellent ee were obtained in most cases.
Xu and co-workers have recently developed a Rh(I)-diene catalytic system able to exploit water as proton source for the asymmetric hydroarylation of α-aminoalkyl acrylates (45) with boronic acids [34][83]. In the same year, Xia’s group developed an efficient protocol using a well-designed chiral Rh-diene catalyst with arylboronic acids and alkenylboronic acids in the enantioselective hydroarylation/hydroalkenylation of benzo[b]thiophene 1,1-dioxides (47) [35][84]. This protocol is free of base and additive, and it uses water as co-solvent. Structures of employed chiral ligands were in Figure 14 together with scheme of reactions.
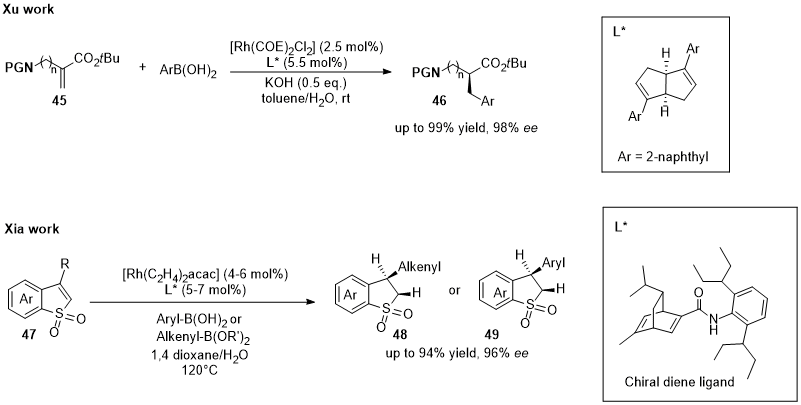
Figure 14. Diboron–water species as hydride source for the Rh-catalyzed asymmetric synthesis of α-aminoalkyl acrylates (46) (by Xu) and benzo[b]thiophene 1,1-dioxides (48 or 49) (by Xia).