1. Immunosuppressive Cytokine Pathways
Regulatory T-cells (Treg) are a subpopulation of suppressor CD4+T-cells that are involved in the suppression of immune-mediated tissue destruction towards self-antigens, helping prevent the development of autoimmunity
[1][42]. However, Treg cells support tumor progression by secreting inhibitory cytokines such as Transforming Growth Factor-β (TGFβ), Interleukin-10 (IL-10), and IL-35 inhibiting the anti-tumor immune response of effector T cells, natural killer (NK) cells and dendritic cells (DCs) resulting in tumor progression
[2][43]. Targeting these immunosuppressive cytokines overcomes ICI resistance by increasing effector T cells and NK cell infiltration and by decreasing the myeloid-derived suppressor cell (MDSC) population within the TME
[1][42]. It is therefore hypothesized that the modulation of certain cytokines, such as chemokine ligand 12 (CXCL12), TGFβ, and IL-17A, using monoclonal antibodies could be an avenue to potentiate the efficacy of PD-1/PD-L1 blockers by surpassing primary or acquired resistance in these therapy-refractory cancers
[3][4][5][6][44,45,46,47].
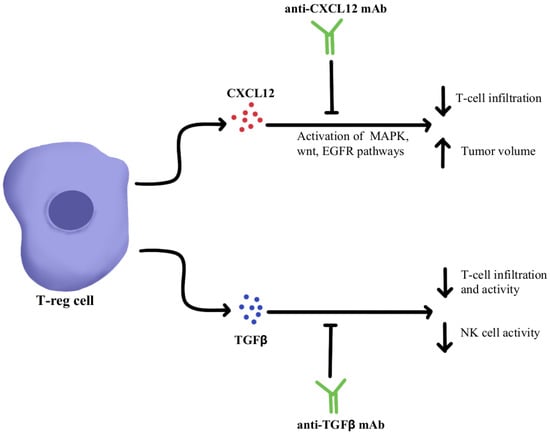
CXCL12 is a cytokine that is localized in tumor cells and binds to chemokine receptor 4 (CXCR4) to activate multiple signaling pathways, including mitogen-activated protein kinase (MAPK), wingless-related integration site (wnt), and epithelial growth factor/receptor (EGF/EGFR) for cell cycle progression and migration
[7][8][48,49]. Through utilizing an antagonist to inhibit CXCL12 binding to CXCR4 in pancreatic cancer murine models, one study showed a marked increase in effector T-cell accumulation in the tumors and a reduction of tumor cell volume despite the continued presence of Treg cells. These results suggest that by neutralizing CXCL12 signaling, Treg-induced inhibitory pathways in tumors can be bypassed, allowing the recruitment of T-cells and thereby re-sensitizing immune cells and tumor cells to PD-1/PD-L1 checkpoint inhibition, as illustrated in
Figure 1 [7][48]. Similar results were seen in pre-clinical studies where CXCL12 antagonism in metastatic breast cancer led to increased T-cell infiltration and improved immunotherapeutic efficacy in murine models
[9][50]. With the intent of clinical translation, clinical trials have combined CXCL12 antagonist and anti-PD-1/PD-L1 therapies and have shown these treatments to be safe and efficacious in metastatic pancreatic cancer and colorectal cancer
[10][11][51,52]. In these studies, combination therapy consisting of CXCL12 antagonists and ICIs showed synergism and enriched TME with effector T-cells to generate a robust anti-tumor response.
Figure 1. Diagram of CXCL12 and TGFβ cytokine pathway modulation
[7][8][9][10][11][12][13][14][15][16][17][18][48,49,50,51,52,53,54,55,56,57,58,59]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
TGFβ is a cytokine that drives numerous cell processes, ranging from proliferation, motility, apoptosis, differentiation, and immune regulation
[12][53]. TGFβ signaling in the immune TME, as illustrated in
Figure 1, represses the anti-tumor activity of the immune cell population, including T cells and NK cells, resulting in immunosuppression that further limits the efficacy of ICIs and, as a result, induces therapeutic resistance to ICIs
[13][54]. Studies using TGFβ inhibitors have been shown to increase T cell infiltration, Thelper1 (Th1)-type immune response, upregulation of Interferon-γ (IFN-γ), and promoted the lytic function of tumor antigen-specific CD8+T cells
[14][55]. Further studies using a dual approach by simultaneously blocking the PD-1/PD-L1 checkpoints as well as the TGFβ pathway were successful in mounting a significant anti-tumor response by overcoming the ICI resistance as well as enhancing the recruitment of T cells
[14][15][16][55,56,57]. The clinical data of TGFβ pathway inhibitors alone and in combination with ICIs have been studied in multiple advanced treatment-refractory cancers including pancreatic, cervix, colorectal carcinoma, biliary tract cancer, urothelial carcinoma, and NSCLC, and have shown to be safe, well-tolerated and provide better clinical outcomes (NCT03631706, NCT03732274)
[17][18][58,59].
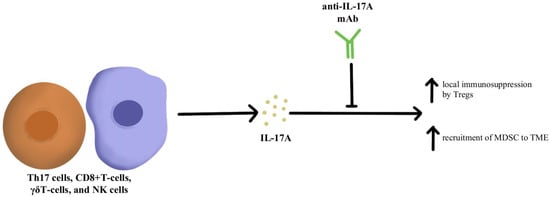
IL-17A is a proinflammatory cytokine that is produced by various cells, including Th17 cells, CD8+T-cells, γδT-cells, and NK cells within the TME
[19][60]. IL-17A is expressed across different tumor types; however, the impact of IL-17A appears to be different based on the type of cancer. For instance, IL-17A serves as a tumor suppressor in melanoma and ovarian cancer, while in other cancers such as colorectal cancer and non-small cell lung cancer, IL-17A supports tumor growth
[4][20][21][22][45,61,62,63]. Looking specifically at the impact of IL-17A and its relationship to checkpoint inhibitors, IL-17A may potentiate the recruitment of MDSC and increase the immunosuppressive activity of Tregs within the TME that ultimately leads to resistance to anti-PD-1/PD-L1 therapy, as illustrated in
Figure 2 [23][24][25][64,65,66]. Pre-clinical evidence suggests that IL-17A can promote migration and invasion of cancer cells by upregulating matrix metalloproteinase, and inhibition of a nuclear factor-kb (NF-kb) abolishes the tumor-promoting effects of IL-17A. These laboratory observations have not yet been translated into clinical applications. Due to the differential effects of IL-17A seen in different cancer types, the exact role of IL-17A in protective and aberrant processes has yet to be fully elucidated; however, this cytokine remains a possible modulation target to immunopotentiate the TME in ICI-refractory tumors
[26][67].
Figure 2. Diagram of IL-17A cytokine pathway modulation [4][19][20][21][22][23][24][25][26]. Illustrated by Kaitlyn Mi of The Dartmouth Institute. Diagram of IL-17A cytokine pathway modulation [45,60,61,62,63,64,65,66,67]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
2. T-Cell Exhaustion and Depletion
T-cell exhaustion within the TME is a significant mechanism of resistance to anti-PD-1/PD-L1 therapy. Exhausted T cells have been found to upregulate multiple inhibitory receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA4), T-cell immunoglobulin, mucin-3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and the T-cell tyrosine-based inhibitory motif (ITIM) domain
[27][28][29][30][31][68,69,70,71,72]. Mutation of the ITIM domain affects PD-1 signaling and T-cell functional activity
[32][73]. Overexpression of the T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) and increased infiltration of CD8+TIGIT+T-cells have been noted as correlating with worse prognosis in several cancers. Studies have shown that dual blockade of TIGIT/PD-1 blockers can overcome the T-cell dysfunction/exhaustion pathway of resistance
[33][74]. CTLA4, discovered by 2018 Nobel Prize co-recipient James Allison, is an immune checkpoint protein expressed on activated T-cells that binds either CD80 and/or CD86 found on antigen-presenting cells (APCs) and regulates T-cell activity and effector functions
[34][75]. CTLA4 and PD-1/PD-L1 pathways interact with each other in cancer in a complex manner, and studies have clearly shown the benefit of combining anti-CTLA4/anti-PD-1 treatments, both in preclinical and clinical settings, to reverse T-cell exhaustion
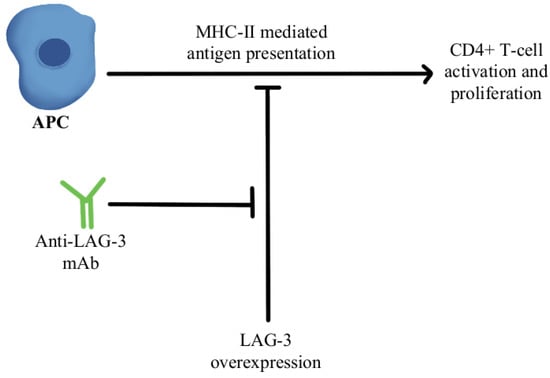
[35][76]. LAG-3 interacts with major histocompatibility-II (MHC-II) to prohibit the binding of MHC molecules to T-cell receptor (TCR) and CD4+T cells, therefore, directly hindering TCR signaling in immune response
[36][77]. A combination of anti-LAG-3 antibodies with nivolumab in a clinical study has shown it to be a safe and efficacious regimen in immunotherapy-resistant melanoma through its modulation of this pathway, as can be seen in
Figure 3 [37][78].
Figure 3. Diagram of LAG-3 pathway modulation [36][37]. Illustrated by Kaitlyn Mi of The Dartmouth Institute. Diagram of LAG-3 pathway modulation [77,78]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
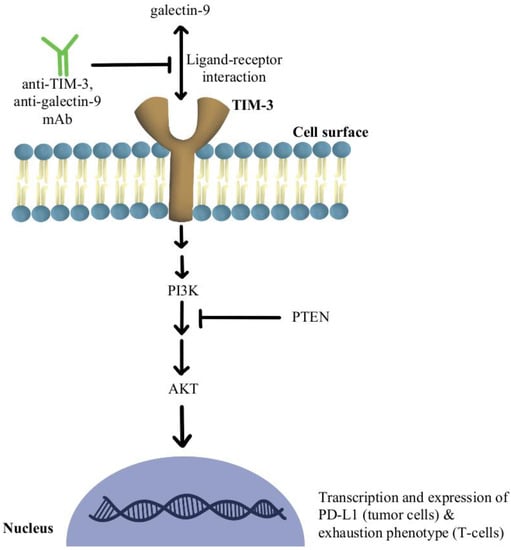
Another significant mechanism of resistance is the upregulation of TIM-3 on dysfunctional tumor-infiltrating lymphocytes (TILs), leading to an exhaustion phenotype
[38][79]. It has been shown that over-expression of TIM-3 on Treg cells is associated with the development of resistance to anti-PD-1/PD-L1 therapy in melanoma
[39][80]. The use of TIM-3 antibody in combination with chemotherapy indirectly enhanced CD8+T-cell responses as well as increased chemokine ligand 9 (CXCL9) expression by DCs
[40][81]. TIM-3 mediates the exhaustion of T-cells and escape of PD-1 inhibition by activating phospho-inositol-3 kinase (PI3K)/protein kinase B(Akt) signaling
[41][82]. In phosphatase and tensin homolog (PTEN) deleted tumors, PI3K/AKT pathway enhances the expression of PD-L1 and inactivates T-cells. Further, a selective PI3Kβ inhibitor in murine cancer models has revealed improvements in the efficacy of both anti-PD-1 and anti-CTLA-4 antibodies
[42][83]. Additionally, a clinical study combining anti-TIM-3 antibody with anti-PD-1 immunotherapy in 219 patients was deemed safe and efficacious in solid malignancies
[43][84]. There is also an ongoing clinical trial, slated to complete in December of 2022, that is investigating the modulation of TIM-3′s ligand, galectin-9, in potentiating the efficacy of anti-PD1 therapy in the treatment of solid tumors (NCT04666688).
Figure 4 contains a diagram of this pathway and its modulation targets.
Figure 4. Diagram of TIM-3 pathway modulation (NCT04666688) [38][39][40][41][42][43]. Illustrated by Kaitlyn Mi of The Dartmouth Institute. Diagram of TIM-3 pathway modulation (NCT04666688) [79,80,81,82,83,84]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
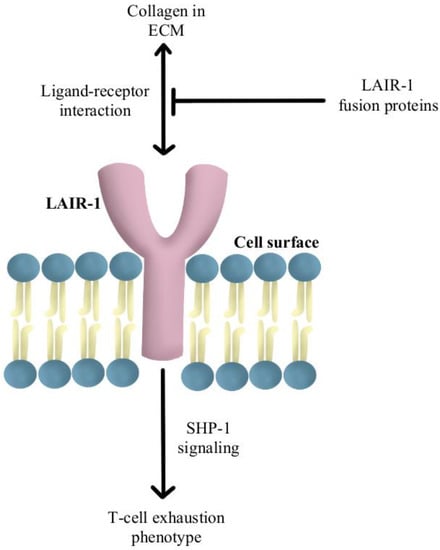
Another reported mechanism of resistance to PD-1/PD-L1 checkpoint inhibitors is increased levels of collagen in the tumor extracellular matrix that activates leukocyte-associated immunoglobulin-like receptor (LAIR-1), a membrane-bound receptor expressed by CD8+T-cells following CD18 interaction with collagen, causing a signaling cascade that induces T-cell exhaustion through SHP-1 signaling
[44][85]. Preliminary in vivo studies have shown that LAIR-2 protein, a soluble receptor with greater affinity for collagen than LAIR-1, regulates the immune system by preventing LAIR-1 cross-linking to collagen via competitive binding
[45][86]. Experiments are currently underway using LAIR-1 fusion proteins to probe for potential ligands for this inhibitory receptor for pre-clinical and clinical use, as diagrammed in
Figure 5 [46][87].
Figure 5. Diagram of LAIR-1 pathway modulation [44][45][46]. Illustrated by Kaitlyn Mi of The Dartmouth Institute. Diagram of LAIR-1 pathway modulation [85,86,87]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
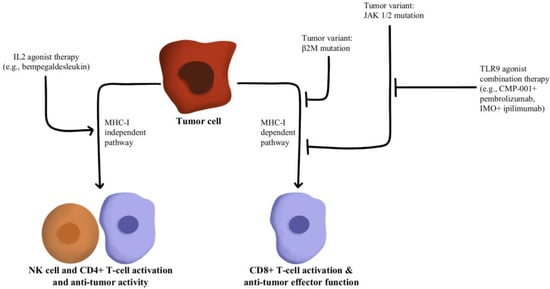
T-cells recognize antigens presented by APCs. However, mutations in beta-2-microglobulin (B2M) can disrupt antigen presentation that ultimately leads to immune checkpoint blockade therapy resistance
[47][48][88,89]. Whole exome sequencing of immunotherapy-resistant tumors has shown that truncating mutations in B2M and mutations in the genes encoding interferon-receptor-associated Janus kinase 1/2 (JAK1/2) result in a lack of response to IFN-γ with loss of surface expression of major histocompatibility complex class I
[49][90]. With this loss of MHC class I, tumors with the B2M mutation respond to anti-PD-1 therapies through the activation of MHC class I-independent mechanisms mediated by CD4+T-cells or NK cells
[50][91]. Bempegaldesleukin, a CD122-preferential IL-2 pathway agonist, has been shown to activate CD4+T-cells and NK cells regardless of the PD-1/PD-L1 status of tumors
[51][92]. Additionally, when Bempegaldesleukin was used in combination with immunotherapeutics, including anti-PD-1 and anti-CTLA4, it resulted in intratumoral depletion of Treg cells and increased proliferation of CD8+T-cells
[52][93]. As the mutations in JAK1/2 led to resistance to PD-1 blockade therapy, a preclinical study has shown that administration of Toll-like receptor 9 (TLR9) agonists can overcome JAK1/2 knockout-induced resistance mediated by NK and CD8+T-cells
[53][94]. Following these observations, CMP-001 (a TLR9 agonist) has been developed for study in a phase Ib clinical study in combination with Pembrolizumab in advanced melanoma (NCT02680184). Another phase II study evaluated the combination of IMO (another TLR9 agonist) in combination with ipilimumab (anti-CTLA4) in immunotherapy-refractory melanoma and has shown a revival of immune responses in 15 out of 24 enrolled patients
[54][95]. These drug combinations and their respective effects on the MHC-I-independent and MHC-I-dependent pathways are illustrated in
Figure 6.
Figure 6. Diagram of MHC-I-dependent and MHC-I-independent pathway modulation
[47][48][49][50][51][52][53][54][88,89,90,91,92,93,94,95]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
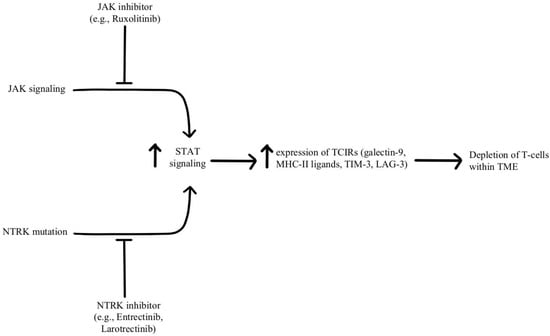
Depletion of T-cells within the TME is facilitated by chronic, persistent type II interferon signaling, enabling signal transducer and activator of transcription (STAT) tumor-related epigenetic changes, resulting in increased expression of interferon-stimulated genes and inhibitory receptors (TCIRs) on multiple T-cells, including Galectin-9, MHC-II ligands, and immune inhibitory checkpoints, including TIM-3 and LAG-3. Increased co-expression of multiple TCIRs aggravates T-cell depletion, while blocking interferons can reverse resistance caused by T-cell depletion
[55][56][96,97]. Konen and others have found that neurotrophic tyrosine receptor kinase (NTRK) is upregulated by anti-PD-1 therapy. NTRK also activates the JAK-STAT signaling pathway and by upregulating the expression of multiple inhibitory receptors on T-cell surfaces, promoting T-cell exhaustion, as illustrated in
Figure 7 [57][98]. There are currently two FDA-approved drugs indicated for the treatment of solid tumors with NTRK gene fusion, entrectinib, and larotrectinib, which act through selective inhibition of the tyrosine kinase domain of the NTRK
[58][4]. Patients experiencing resistance to ICI therapy may see therapeutic benefits with concurrent administration of one of these small molecule inhibitors due to its attenuation of that JAK-STAT signaling pathway and prevention of T-cell exhaustion, however, the repurposing of these drugs for treating ICI resistance remains an open avenue of investigation.
Figure 7. Diagram of STAT pathway modulation [55][56][57][58]. Illustrated by Kaitlyn Mi of The Dartmouth Institute. Diagram of STAT pathway modulation [4,96,97,98]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
3. Adoptive Cellular Therapy
Chimeric antigen receptor T-cell (CAR-T) therapy has been shown to achieve durable remissions in cancers and is currently FDA-approved for the treatment of certain hematological malignancies, and its combination with checkpoint inhibitors targeting PD-1 has provided successful outcomes in several cancers such as B-cell non-Hodgkin lymphoma, hepatocellular carcinoma, and metastatic melanoma
[59][60][61][62][63][99,100,101,102,103].
In a safety and efficacy clinical trial, the use of anti-CD19 CAR-T cells in combination with nivolumab in 11 patients with relapsed/refractory non-Hodgkin lymphoma has shown that this combined treatment regimen is safe and mediates potent anti-lymphoma activity
[64][104]. Another study utilized HER-2+ transgenic mice models to study the effects of CAR-T cells with and without PD-1 inhibition, and it was observed that the combination therapy improved the therapeutic efficacy of CAR-T-cells against solid cancers
[65][105]. The synergy seen in the combination therapy is likely multifactorial, being a product of increased T-cell effector activity and other effects, such as enhanced T-cell survival
[66][106].
Investigations into the gene editing of CAR-T cells have revealed that PD1-knockout CAR-T cells are a clinically efficacious alternative to combination therapy. Specifically, it was observed in murine models that editing the PD1 gene on tumor-specific T-cells to reduce the expression of PD-1 resulted in a significant delay in tumor growth following administration to PD-L1 overexpressing tumors
[67][68][107,108]. Further data supports these findings, in that PD-1 deficient CAR-T cells showed significantly better antitumor efficacy than wild-type CAR-T cells
[69][70][71][109,110,111].
In pursuit of more effective treatments, the engineering of CAR-T cells has been explored, such as the modification of CAR-T cells to secrete PD-1 blockers–a modification that was found to control tumor growth in lung cancer models significantly better than standard CAR-T cell therapy
[72][112]. In a similar study looking at humanized mice models of renal cell carcinoma, carbonic anhydrase-targeting CAR-T-cells engineered to secrete PD-L1 antibodies were shown to combat T-cell exhaustion and increase recruitment of NK cells to the tumor tissue, while in another study CAR-T cells were engineered to express an anti-PD-1 single chain variable fragment, which was shown to attenuate the PD-1-dependent inhibition of CAR-T cells as well as tumor-specific non-CAR-T cells, resulting in significant control of tumor growth when compared to wild type CAR-T-cells
[73][74][113,114].
In addition, there are several ongoing clinical trials using CAR-T-cell therapy in combination with checkpoint blockers in the treatment of B-cell lymphoma, multiple myeloma, and Hodgkin lymphoma (NCT04134325, NCT04162119, NCT04163302, and NCT04213469). CAR-T-cell therapy and PD-1 inhibitors have both individually revolutionized the field of oncology, and their combination has been shown to be both synergistic and efficacious in overcoming resistance to checkpoint inhibition.
4. Tumor Neoantigen Vaccines
Heterogeneous response to PD-1/PD-L1 immunotherapy has prompted the investigation into strategies to overcome primary and acquired resistance. One such polypharmaceutical approach is the use of tumor neoantigen vaccines in combination with ICIs to potentiate the tumor-specific immune responses
[75][76][115,116].
Tumor antigens, or neoantigens, are proteins displayed by tumor cells due to mutations in host DNA. Neoantigens stimulate CD4/8-MHC cell-mediated immune reactions, but clonal evolution promotes evasion of the normal response
[77][117]. Studies support that tumor neoantigen vaccination is effective in individuals with an absence of pre-existing antitumor immunity. However, the TME expresses several immunosuppressive factors through prolonged exposure to cancer antigens, and/or poor infiltration of T-cells
[78][118]. Anti-PD-1/PD-L1 immunotherapy has shown promising results in many solid tumors, with pancreatic cancer and glioblastomas being a few exceptions, possibly due to the lack of permeability of T-cells in tumor tissue
[79][119]. A study outlined the following 3 distinct phenotypes describing the permeability of tumor cells based on epigenetic RNA N6-methyladenosine (m6A) modification in 1938 gastric cancer samples: immune-excluded, immune-inflamed, and immune-desert phenotypes. Based on their study, a low m6A score (indicating low levels of methylation and greater T-cell tissue infiltration) predicted an immune-inflamed phenotype, a better response to PD-1/PD-L1 immunotherapy, and a 5-year survival rate of 69.4%
[80][120]. Cancer vaccines targeting neoantigens have been proposed as a tool to circumvent immunotherapy-induced resistance, increase T-cell permeability, and decrease T-cell exhaustion, potentially increasing the effectiveness of ICIs
[78][118].
A Phase 1b feasibility study used the DC neoantigen vaccine in combination with anti-PD-1 therapy in pancreatic ductal adenocarcinomas to reprogram the TME and showed positive outcomes, a step forward in using these advanced therapeutic platforms, especially for treatment-refractory cancers
[78][118]. Another vaccine approach currently in studies is using PD-1/PD-L1 checkpoint inhibitors with the GVAX vaccine–an anti-cancer vaccine consisting of irradiated tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF)
[81][121]. GM-CSF is a hematopoietic cytokine responsible for inducing the production of white blood cells, including granulocytes, macrophages, and megakaryocytes. GM-CSF also functions as a major pro-inflammatory cytokine, coordinating communication between lymphocytes and myeloid cells
[82][122]. Another study combined PD-1/CTLA-4 checkpoint blockade with the GVAX vaccine in preclinical models of colon cancer and ovarian cancer with the eradication of tumor cells
[83][123]. In addition to the GVAX vaccine, GM-CSF is used in the similar yet distinct treatment modality of oncolytic virotherapy, appearing as one of the principal genetic modifications in the only current FDA-approved oncolytic virus, Talimogene laherparepvec (T-vec), wherein GM-CSF likewise serves a proinflammatory role, potentiating an anti-tumor immune response. T-vec has shown clinical efficacy in treating metastatic melanoma, for which it received its original FDA approval, while recent investigations into its combination with PD-1 immunotherapy have shown promising clinical benefits
[58][4]. This success has further galvanized interest in the combination of oncolytic viruses with PD1 inhibitors, with the field seeing an explosion in the diversity of viruses, spanning both DNA and RNA viruses, used in combination with PD1 checkpoint therapy
[84][124].