Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fabio Penna and Version 2 by Conner Chen.
Cancer metabolism is an emerging field of investigation aimed at identifying cancer cell vulnerabilities in order to define novel anti-cancer therapeutic approaches based on interventions that modulate the availability of specific nutrients. Amino acids (AAs) are used by cancer cells as both building blocks for protein synthesis required for rapid tumor growth and as sources of energy.
- amino acid
- cancer metabolism
- cachexia
- nutrition
1. Impact of AAs on Cancer Metabolism and Mitochondrial Function
The elevated biosynthetic activity of cancer cells is supported by several strategies aimed at increasing growth. Oncogenic mutations allow tumor cells to sustain angiogenesis, evade apoptosis, enable constitutive proliferation signaling by tyrosine kinase receptors, and to escape from growth suppression signaling [1][2]. Moreover, profound metabolic alterations also confer to cancers the ability to optimize substrate utilization; one typical example of metabolic reprogramming is the Warburg effect (aerobic glycolysis), the observation that cancer cells convert glucose to lactate even in an oxygen-rich environment, dates back to almost 100 years ago [2][3]. The Warburg effect is not exclusive to cancer cells, but also occurs in normal, non-cancerous, rapidly dividing cells, such as activated macrophages and lymphocytes, haematopoietic stem and progenitor cells, or during angiogenesis, and this confirms the growth advantage conferred by this metabolic adaptation [3][4]. Initially ascribed to defective mitochondria, this apparently inefficient metabolic reprogramming is now hypothesized to have the aim, rather than ATP production, of maximizing carbon delivery to the cell’s anabolic pathway for the synthesis of biomass. In both living organisms and in culture, cells in fact rarely experience shortages of glucose, which is always kept at high/constant levels in the culture medium or bloodstream; this means that proliferating cells do not have a real need to maximize ATP synthesis from glucose [4][5]. Therefore, by engaging glucose into aerobic glycolysis, cancer cells avoid its complete catabolism to ATP in mitochondria; this spares glucose carbon that can be used for generating acetyl-Coa, glycolytic intermediates, and ribose for the synthesis of fatty acids, non-essential amino acids (NEAA), and nucleotides, respectively [5][6]. Since the major part of these pathways require functional mitochondria, this implies that, in proliferating cancer cells, mitochondria are not dysfunctional; however, as a consequence of aerobic glycolysis, they are rather used as a biosynthetic organelle for the synthesis of glucose-derived lipid and NEAA, instead of ATP. Furthermore, cancer cells are also able to efficiently utilize mitochondria for directing other cell substrates for macromolecule synthesis and, in this process, amino acids (AA) play a pivotal role [6][7]. The role of AAs as constituents of proteins and/or signaling molecules involved in the regulation of macroautophagy, the process of endosomal/lysosomal recycling of cellular components, or as activators of biosynthetic cell pathways through mammalian target of rapamycin (mTOR), has been the subject of many other comprehensive reviews [7][8][9][8,9,10]. However, AAs show an intimate connection with mitochondrial function. One such example is glutamine (Gln), which, together with glucose, is the main molecule utilized by the majority of mammalian cells in culture.
2. Cancer Mitochondria Support Biosynthesis of Macromolecules through Glutamine-Dependent Anaplerosis
Since the early discovery that HeLa cells consume Gln from 10 to 100 orders of magnitude more than other amino acids [10][11], and that Gln is used as a major source of cell energy, rather than for incorporation into proteins [11][12], the observation that transformed cells display a high rate of Gln consumption has been confirmed in several other cancer cell types, such as glioblastoma, ovarian, pancreatic, and breast cancer [12][13][14][15][13,14,15,16]. Through mitochondrial glutaminolysis, Gln is utilized to provide both carbon and nitrogen for anabolic reactions; in particular, through the process of anaplerosis, which leads to the replenishment of the pools of metabolic intermediates of the tricarboxylic acid cycle (TCA) in times of high energy requirements [16][17], Gln is a major carbon source for the synthesis of proteins, nucleotides, and lipids. In this pathway, Gln is first converted into glutamate (Glu) via mitochondrial glutaminase (GLS), and then glutamate dehydrogenase (GDH) convert glutamate into α-ketoglutarate (α-KG); alternatively, a second pathway of conversion of Glu in α-KG involves the activity of both mitochondrial and cytosolic transaminases such as aspartate transaminase (AST), alanine transaminase (ALT), or phosphoserine transaminase (PSAT). The routing of glutamate to the dehydrogenation or to the transamination pathway depend on the metabolic status of the cell and has important metabolic consequences; although the prevalence of transamination over dehydrogenation has been positively correlated with proliferation rate [17][18] and glucose levels, both pathways enable the optimization of amino acid uptake and consumption with growth and biomass production, with transamination being prevalent in times of glucose abundance, while the glutamate dehydrogenation pathway has been shown to be induced during glucose shortages, thus enabling cell survival [18][19][19,20]. Furthermore, sequential glutaminase and glutamate dehydrogenase reactions produce ammonia (NH3) and ammonium (NH4+), respectively, which is considered a cell toxic by-product which requires scavenging through urea; however, ammonia is also a major diffusible autophagy inducer [20][21], which could therefore help cancer cells increase fitness by eliminating damaged and potentially toxic macromolecules and organelles. Moreover, in breast cancer, extracellular ammonia synthesized in the liver can be recycled by tumor cells through reductive amination by GDH to form glutamate which is then used for macromolecule biosynthesis [21][22], and Gln-derived ammonia can also promote sterol regulatory element binding protein (SREBP) mediated lipogenesis and support tumor growth [22][23]. A graphical representation of the above-described metabolic scenario is provided in Figure 1. Glutamate transamination, on the contrary, does not produce ammonia, but generates the NEAAs aspartate (Asp), alanine (Ala), and serine (Ser) which, together with α-KG, serve as anaplerotic substrates in the TCA cycle, thus promoting biosynthesis. In this process, Oxaloacetate (Oaa) produced by α-KG from glutaminolysis condenses with pyruvate-derived Acetyl-CoA to produce citrate at the citrate synthase step; citrate is then not committed to the isocitrate dehydrogenase step (which is instead involved in reductive carboxylation—see below), but is instead exported to cytosol where ATP-citrate lyase reforms Acetyl-CoA, which is then used for malonyl-Coa synthesis and de novo lipogenesis. Glutaminolysis flux also generates reduced nicotinamide adenine dinucleotide (NADH) and reduced nicotinamide adenine dinucleotide phosphate (NADPH), as reducing equivalents for the electron transport chain (ETC) and both redox balance and lipogenesis, respectively. Beyond glutaminolysis, which is considered as the oxidative branch of Gln metabolism, the alternate metabolic fate of Gln is through the reductive carboxylation pathway, which involves the engaging of α-KG in a “reverse” mitochondrial TCA cycle through NADPH-dependent isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) and therefore producing citrate for lipid biosynthesis [23][24][25][24,25,26]. This pathway seems to be predominant in cancer subjected to hypoxia, displaying constitutive hypoxia-inducible factor 1 alpha (HIF1α) activation, or have defective mitochondria [26][27][27,28] and has been confirmed to occur also in vivo [27][28][28,29].
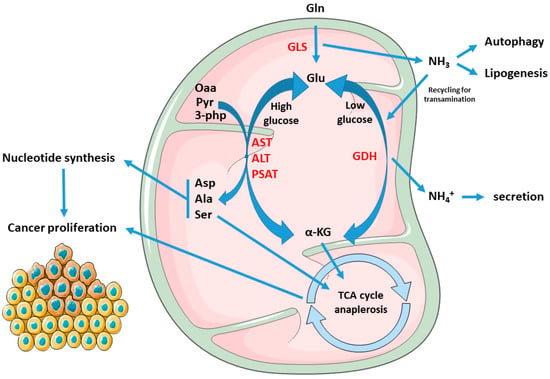
Figure 1. Rewiring of the metabolic fate of AAs in cancers: Gln- derived Glu yield α-kg through both transamination or dehydrogenation, by means of AST, ALT, PSAT, or GDH, respectively. The first pathway prevails in conditions of glucose abundance, resulting in production of the NEAAs Asp, Ala, and Ser, and enabling proliferation. Asp is used in nucleotide synthesis while α-kg, Ala, and Ser can feed the TCA cycle. On the other hand, GDH is activated during glucose scarcity or cell quiescence, supporting cell survival and stress resistance. Furthermore, GLS-derived ammonia in cancer can stimulate autophagy, lipogenesis, or being re-incorporated to Glu synthesis, which can be re-routed for transamination and biomass production. Abbreviations: Gln, glutamine; Glu, glutamate; Asp, aspartate; Ala, alanine; Ser, serine; GDH, glutamate dehydrogenase; GLS, glutaminase. AST, aspartate transaminase; ALT, alanine transaminase; PSAT, phosphoserine transaminase; Oaa, oxaloacetate; Pyr, pyruvate; 3-Php, 3-phosphohydroxypyruvate; α-KG, alpha-ketoglutarate.
3. Mitochondrial Electron Transport Chain and AA Metabolism
Beyond the evidence coming from glutamine/glutamate metabolism, several other studies clearly demonstrate that the function of mitochondria in sustaining cell proliferation goes beyond the simple ATP synthesis, and that biomass production and macromolecule synthesis depend on to the strict connection and crosslink between mitochondrial activity and AA metabolism. A striking example on how mitochondrial function and AA metabolism are reciprocally intertwined comes from two studies which show that the most essential function of the ETC in proliferating cells is to provide electron acceptors to support Asp biosynthesis which, in turn, is required for the synthesis of purine and pyrimidines [29][30][30,31]. By recycling NAD+, ETC provides oxidized cofactor for malate dehydrogenase 2 (MDH2) which, in turn, produces Oaa that is used by mitochondrial aspartate transaminase (GOT2) for the synthesis of Asp. However, when ETC is dysfunctional, the ratio NAD+/NADH decreases, and Asp synthesis is switched to the reductive carboxylation of Gln to citrate which, through ATP-citrate lyase, produces Oaa to drive Asp synthesis. This suggests that, in proliferating cells, the high NAD+/NADH ratio maintained by mitochondrial respiration is essentially used for Asp/nucleotide synthesis, rather than for ensuring constant TCA cycle/OXPHOS function. Treatment of acute myeloid leukemia (AML) cells with IACS-010759, a complex I inhibitor, reduced cell viability, decreased NAD+/NADH ratio, and, among AAs, exclusively reduced Asp levels while increasing Gln utilization, thus confirming that the interconnection between mitochondrial activity, redox state, and Asp synthesis is extended to cancer cells [31][32]. This relationship has also been recently confirmed in vivo; treatment of mouse neuroblastoma xenografts with IACS-010759 affected ETC activity, redox state and Asp metabolism, as well as activation of reductive carboxylation of Gln. In addition, treatment of xenografted mice with metformin, a commercial available antidiabetic drug and complex I inhibitor, dose-dependently inhibited tumor growth, together with a decrease in intratumor NAD+/NADH ratio and Asp levels [32][33][33,34]. The evidence of reprogramming of the cancer mitochondrial function to support Asp synthesis for survival comes not only from studies based on pharmacological inhibition of complex I, but also from genetic disruption of ETC components, which resemble mitochondrial alterations in tumors; inactivating mutations of mitochondrial succinate dehydrogenase (SDH) are frequent in cancers and increase susceptibility to the development of different tumors and, as such, SDH has tumor suppressor functions. Immortalized kidney mouse cells deficient in SDH become addicted to extracellular pyruvate for proliferation, which, through carboxylation to Oaa, is diverted to Asp synthesis. Notably, SDH ablation in absence of pyruvate also decreased the NAD+/NADH ratio, thus confirming that, in cancer cells, mitochondrial function is strictly connected to Asp biosynthesis through the modulation of its redox state [34][35]. Moreover, a very recent paper revealed a close relationship between ETC, redox state, and Gln utilization for biomass production: in 143B cells, respiration inhibited by both mitochondrial DNA (mtDNA) depletion of parental ρ0 cells and chemical ETC inhibition, the reduced mitochondrial NAD+/NADH ratio reversed mitochondrial GOT2 as well as succinate and malate dehydrogenases to promote mitochondrial oxidation of NADH to NAD+; this, in turn, enabled GDH-dependent Gln anaplerosis to support cell proliferation [35][36]. Beyond Asp, mitochondrial NAD+ regeneration has been shown to be essential also for the synthesis of Ser, which is another carbon source for the production of nucleotides; in fact, withdrawal of Ser from culture medium increased sensitivity of melanoma cell lines to the anti-proliferative effects of both rotenone or metformin; furthermore, Ser deprivation depleted cells of purine nucleotides and rotenone treatment further increased their depletion [36][37]. Alteration of Ser synthesis also occurs in response to cells depleted of mtDNA; this causes bioenergetic failure and induction of activating transcription factor 4 (ATF4), a master transcription factor which regulates cell response to AA starvation during the integrated stress response (ISR) [37][38]. mtDNA depleted cells display increased Ser synthesis and decreased Ser consumption, thus reflecting elevated reliance on serine for survival [38][39]. Ser, together with glycine (Gly), is also involved in one-carbon (1C) metabolism, a process shared by both mitochondria and cytosol which, through the folate cycle, leads to the production of 1C methyl units for several biochemical pathways such as purine and pyrimidine biosynthesis, and synthesis of methionine (Met), as well as providing the moiety for methylation reactions for epigenetic control of gene expression [39][40]. Being a central hub for a plethora of anabolic pathways, cancer cells can therefore become reliant on generation of 1C units from both Gly and Ser. Ser can be converted to Gly by the cytosolic or mitochondrial serine hydroxymethyltransferase SHMT1 and SHMT2, respectively, with the transfer of Ser-derived 1C unit to tetrahydrofolate (THF), generating methylene-THF which, in turn, is required for purine and pyrimidine biosynthesis. As a result, cancer cells have been shown to both avidly consume extracellular Ser and also to strongly depend on endogenous Ser synthesis, displaying an elevated Ser flux [40][41][42][43][41,42,43,44]. Ser can be synthesized from the glycolytic intermediate 3-phosphoglycerate (3-PG) through the enzyme phosphoglycerate dehydrogenase (PHGDH), which is in a genomic locus of copy number gain in both breast cancer and melanoma, and whose protein levels are increased in 70% of estrogen receptor (ER)-negative breast cancers [41][43][42,44]. The overexpression of PHGDH in tumors allows elevated rates of de novo Ser biosynthesis which, as a result, increases the biosynthesis and metabolic pathways associated with the folate pool, amino acid, lipids, and redox regulation [43][44]. However, in tumors, this metabolic adaptation requires a diminished flux of pyruvate toward mitochondrial oxidation, thus redirecting upstream glycolytic intermediates, such as 3-PG, to Ser and Gly synthesis; in many cancer cells, this is accomplished by preferential expression of the M2 isoform of pyruvate kinase (PKM2), which displays lower kinase activity compared to PKM1, thus promoting the accumulation 3-PG and other glycolytic intermediates which are precursors for biosynthesis of Ser, as well as nucleotides, amino acids, and lipids required for proliferation [44][45]. Notably, Ser is an allosteric activator of PKM2, and this allows cancer cells to rapidly increase Ser biosynthesis in response to its environmental shortage since, when Ser levels fall, the decrease in PKM2 activity allows more glucose-derived carbon to be diverted into serine biosynthesis [45][46]. Although Ser can be synthesized by both SHMT1 and SHMT2, the mitochondrial isoform is strongly upregulated in cancers and has been shown to have a stronger impact on cancer metabolism [41][46][42,47]. Mitochondrial 5,10-methylenetetrahydrofolate production by SHMT2 provides methyl groups for mitochondrial tRNA modification, which are required for proper mitochondrial translation and function [47][48][48,49]. Furthermore, SHMT2 is required for complex I assembly, and mice deficient in SHMT2 display embryonic lethality and defective mitochondrial respiration [49][50][50,51].
4. Targeting AA Metabolism as Anti-Cancer Strategy
The data herein reported indicate that the connection between AA metabolism and cancer mitochondrial function goes beyond the well-known role of AAs as mitochondrial substrates and can also explain why tumors are dependent on some AAs. Notably, this addiction also includes AAs usually classified as non-essential such as Gln, Ser, and Asp, therefore implying that the mitochondrial reprogramming of cancer metabolism also re-defines and crosses the distinction between essential and non-essential AAs. This AA dependency highlights a metabolic vulnerability, which could be exploited for a highly specific anticancer therapy aimed to starve or deplete cancer cells of selected amino acids, or to block crucial AA metabolic pathways [51][52].
GLN Glutaminase inhibition has been long proposed as a main cancer therapeutic target, and several Glnase inhibitors have been developed, of which the most studied are 6-diazo-5-oxo-L-norleucine (DON), bis-2-(5-phenyl acetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES), and CB-839; however, since glutaminolysis is not an exclusive feature of cancer cells (see above), low specificity/high toxicity concerns and poor solubility issues have limited their employment in clinic, and only CB-839 (Telaglenastat) has since been entered in a clinical trial and is currently in phase II [52][53].
ASN Asparagine (Asn) depletion by means of the bacterial enzyme asparaginase (ASNase) is presently the only approved anticancer treatment based on an AA-depleting approach and has been long used as an anticancer strategy, especially toward pediatric acute lymphoblastic leukemia (ALL) [53][54][54,55]. Contrary to most cells, leukemia cells express low levels of asparagine synthetase (ASNS), which render them highly dependent on Asn, thus making ASNase treatment effective [55][56]. Although several toxic side effects have also been also reported, the majority of them are manageable, but ASNase is almost exclusively used only in ALL [56][57].
ARG Inhibition of cancer cell proliferation by means of arginase (ARGase) treatment was reported almost 70 years ago [57][58]; since then, the depletion of Arginine (Arg) through ARGase or arginine deiminase (ADI) administration has been constantly explored as an anticancer therapy, also supported by the finding of absent or low expression of argininosuccinate synthetase (ASS1) in several tumors, especially those associated with chemoresistance and poor clinical outcome, such as hepatocellular carcinoma (HCC), melanoma, mesothelioma, pancreatic cancer, prostate cancer, renal cell carcinoma, sarcoma, and small cell lung cancer [58][59][60][61][59,60,61,62], which result in Arg dependence. While Arg-deprived, non-cancer cells undergo quiescence and cycle arrest at G0/G1 phase, cancer cells starved for Arg continue instead to DNA synthesis, leading to unbalanced growth and ultimately to cell death [62][63]. Currently, ARGase, as a PEGylated derivative, is employed in clinical trials for the treatment of HCC, melanoma, prostate adenocarcinoma, and pediatric acute myeloid leukemia (AML), while trials with PEGylated ADI are ongoing for the treatment of small cell lung cancer, melanoma, AML, HCC, and mesothelioma [63][64].
SER, GLY The importance of targeting Ser, Gly, and Met metabolic pathways, which are strictly interconnected to folate 1C metabolism, is highlighted by the long-known use of antifolate drugs as chemoterapics [64][65]; methotrexate (MTX), the most used antifolate, deplete cells of tetrahydrofolate and is routinely used for the treatment of multiple cancers. However, MTX treatment has some concerns of toxicity [65][66]. Since, in tumors, the majority of 1C units derive from Ser, many efforts to develop inhibitors of Ser biosynthesis to inhibit cancer 1C metabolism, alone or in combination with other antifolates, have been put forward [66][67]. Pyrazolopyran, an herbicide compound, which was originally shown to inhibit plant SHMT, has been shown to inhibit human SHMT1 and to induce cell death in lung cancer cells [67][68]. An optimized pyrazolopyran derivative, by targeting both SHMT1 and SHMT2, blocked proliferation of colon, pancreatic, and several B-Cell derived malignancies, which displayed different grades of sensitivity according to their proficiency in folate metabolism and glycine uptake, thus confirming amino acid vulnerability as a key target for anti-cancer drug development. Furthermore, incubation of many cancer cells in medium lacking Ser/Gly greatly increased the sensitivity of the cells to the PHGDH inhibitor PH755, while PH755 treatment in vivo to xenografted mice significantly potentiated the anti-cancer response of dietary serine/glycine restriction, thus underscoring the effectiveness of a combined dietary and chemotherapeutic approach [68][69].