Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Rakesh Trivedi.
Many proteins and protein segments cannot attain a single stable three-dimensional structure under physiological conditions; instead, they adopt multiple interconverting conformational states. Such intrinsically disordered proteins or protein segments are highly abundant across proteomes, and are involved in various effector functions.
- protein structure
- protein function
- intrinsically disordered proteins
- intrinsically disordered regions
1. Introduction
The functional aspects of genes have been attributed to RNAs and proteins. Of the two, proteins are the ones that bring about the majority of diverse cellular effector functions. Two paradigms concerning the structure and function of proteins have evolved, as shown in Figure 1 [1,2,3,4,5,6][1][2][3][4][5][6]. The first paradigm corresponds to the well-established, often assumed as the default for proteins, ‘structure–function paradigm’, which states that the three-dimensional native structure under physiological conditions is the prerequisite for a protein to function. The second paradigm is the recently established ‘disorder–function paradigm’ based on the proteins that perform cellular functions without attaining a stable three-dimensional structure under physiological conditions [7]. The naturally occurring, biologically active proteins that appear to possess a high degree of conformational flexibility have been referred to as intrinsically disordered proteins (IDPs) [5,8,9,10][5][8][9][10]. In most instances, instead of the whole protein, only some regions in the protein are disordered and functional; such protein segments are known as intrinsically disordered regions (IDRs) [11,12,13,14][11][12][13][14]. Interestingly, these intrinsically disordered proteins/regions have endowed proteins with functional promiscuity [15,16,17,18][15][16][17][18].
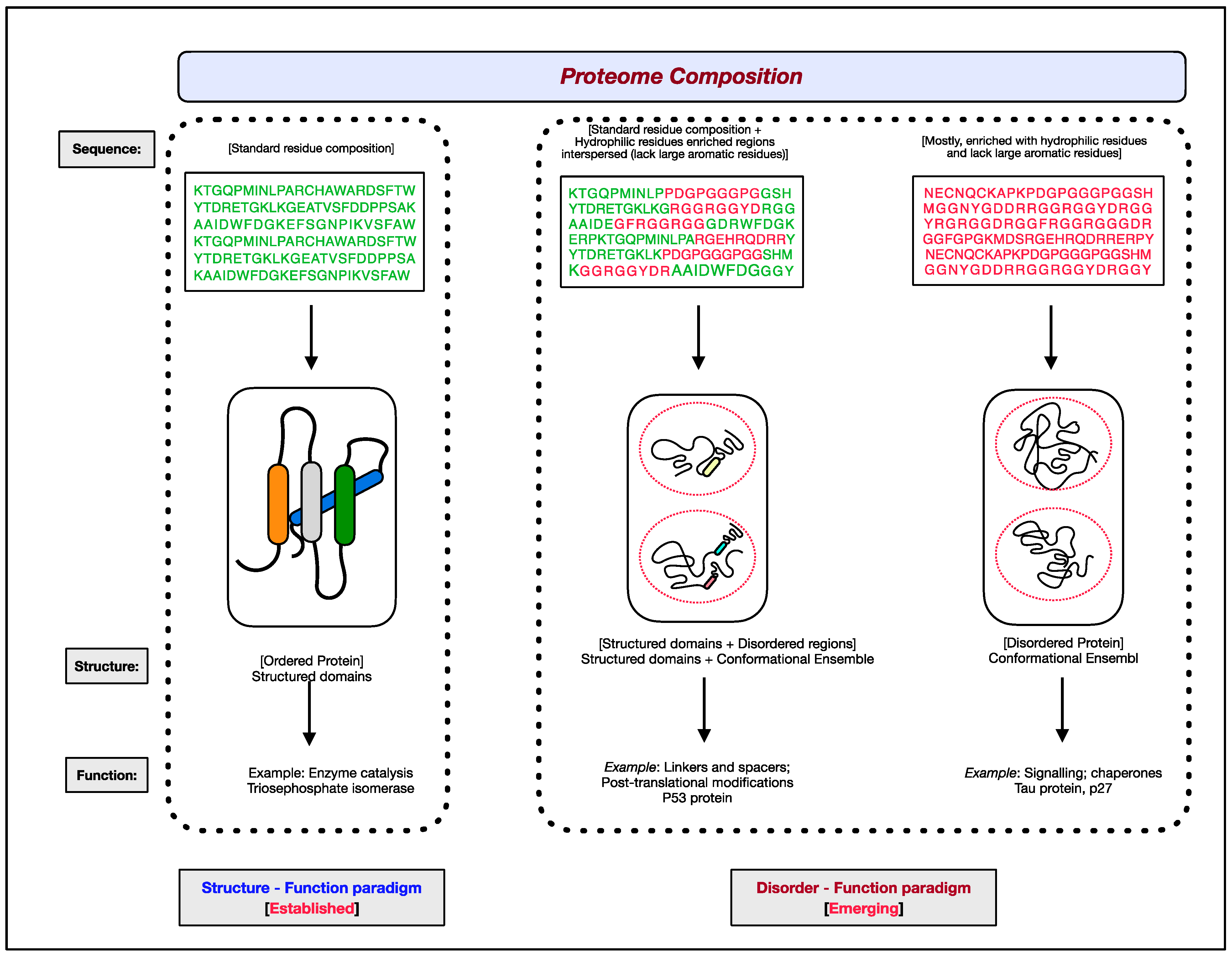
Figure 1. The two paradigms of protein structure and function. According to the well-established ‘structure–function paradigm’, a three-dimensional native structure under physiological conditions is vital for a protein to perform its biological function (for example, enzyme-catalyzed reactions). The more recent ‘disorder–function paradigm’ states that a protein can carry out its biological function without attaining a 3-D stable folded structure under physiological conditions (for example, protein binding to other cellular molecules). For representative purpose, residues coding for ordered/globular domains are shown in ‘green’ color, and residues coding for disordered proteins/segments are shown in ‘red’. At the proteome level, the structured domains and intrinsically disordered regions (IDRs) are two functional building blocks of proteins.
2. Intrinsic Protein Disorder
Repeated occurrences of proteins with intrinsic flexibility and properties different from those of ordered/globular proteins gradually resulted in the development of the notion that non-rigid proteins are not exceptions. Around 2000, these naturally flexible proteins were accepted as a general category of proteins [27,28,29,30][19][20][21][22]. The conformational flexibility of these “non-traditional” proteins was proposed to be the source of their biological functions [31][23]. Over the years, various terms have been introduced by different authors to describe these proteins with inherent flexibility. It was only in recent years that the phrase “intrinsically disordered proteins” (IDPs) became more widely used than other terms [28][20]. Most acceptably, “intrinsic protein disorder” defines the biologically active proteins or protein segments that exist as ensembles of unfolded, collapsed, extended, non-globular conformations at the secondary or tertiary structural level [27,28,29,30,32,33][19][20][21][22][24][25].
2.1. Natural Abundance of Intrinsically Disordered Proteins
Information about IDPs was very sparse for a very long time. Until the reports on the experimentally characterized IDPs, it appeared improbable to have such a class of proteins in abundance [34][26]. However, computational predictions revealed the possible widespread occurrence of IDPs and IDRs [35,36,37][27][28][29]. The exhaustive analysis of 31 genomes spanning three kingdoms of life revealed that a considerable number of proteins contain regions with 40 or more consecutive disordered residues [28][20]. The proportion of structural disorder was also found to increase progressively with genome complexity, from bacteria to archaea, and then to eukaryotes [28,38][20][30]. While 33% of eukaryotic proteins have been reported to contain at least one functionally relevant long (>30 residues) intrinsically disordered region, archaean and eubacterial proteins possess only 2.0% and 4.2% of such functional IDRs, respectively [36][28]. In viral proteomes, the total intrinsic disorder content is determined by the nature of the nucleic acid constituting the viral genome, and it decreases successively with an increase in the size of the viral proteome [39][31]. Recently, 3133 unique proteins were experimentally validated to contain functional long disordered regions (at least 30 residues) [40][32]. Additionally, the degree of disorderedness in proteomes and essential proteins was estimated for various genomes, and a sharp increase was observed at the prokaryote/eukaryote boundary [41][33]. Thus, the natural abundance of disordered proteins or protein segments across complex genomes suggests that, even though IDPs/IDRs fail to attain stable three-dimensional structures under physiological conditions, they are of high functional pertinence [27,28,29,30,42,43,44,45,46,47][19][20][21][22][34][35][36][37][38][39].
2.2. Sequence Characteristics of Intrinsically Disordered Proteins
The propensity of a protein or protein segment to fold or remain unfolded under physiological conditions is encrypted in its amino acid sequence [28,30,32,33,45,46][20][22][24][25][37][38]. In other words, the amino acid sequence composition determines whether a protein would be an ordered protein with a stable folded 3-D structure or an unfolded intrinsically disordered protein. Strong electrostatic repulsions due to a higher net charge and a lack of driving force for compaction due to low mean hydrophobicity are generally considered as the prime reasons for the unfolded, extended structure of IDPs/IDRs [45][37].
An in-depth comparative analysis of the sequence composition of ordered and disordered proteins revealed that residues such as Ala, Arg, Gly, Gln, Glu, Lys, Pro, and Ser (referred to as disorder-promoting residues) occurred more frequently in IDPs/IDRs. In contrast, residues such as Asn, Cys, Ile, Leu, Phe, Val, Trp, and Tyr were more common in the ordered/structured segments of the proteins (referred to as order-promoting residues) [28,48,49,50][20][40][41][42]. Comparative studies of amino acid residues in disordered and ordered regions, physicochemical property-based scales (such as the coordination number, aromaticity, strand propensities, flexibility index, volume, helix propensities, etc.), and composition-based features (e.g., any combination that has one to four residues in the group) have led to the distinction between disordered and ordered regions in proteins [28,50][20][42]. Compositional bias and sequence characteristics play a significant role in defining the interactions of disordered proteins/regions [51,52][43][44]. Furthermore, the distinct compositional bias of the intrinsically disordered proteins/regions as compared with the ordered proteins/regions forms the basis for developing many disorder-predicting computational tools. For instance, tools involving the alignment of IDPs/IDRs use specific amino acid substitution scoring matrices reflecting the frequency of occurrence of different residues in the disordered regions of proteins [53,54,55,56][45][46][47][48].
2.3. Structural Aspects of Intrinsically Disordered Proteins
In general, most proteins exist as a combination of both ordered and disordered segments in different proportions [57,58,59,60,61,62][49][50][51][52][53][54]. Unlike the ordered regions of proteins, which exist as stable secondary/tertiary structures, the disordered regions fail to attain a stable three-dimensional native structure under physiological conditions. However, the structure of IDPs/IDRs can be best defined as the ensemble of functionally relevant interconverting transient structures on a fast time scale.
Several previous studies reported that IDPs/IDRs are enriched with uncharged and polar amino acids, lacks bulky hydrophobic residues, and exist as dynamic heterogeneous ensembles of collapsed or extended structures [63,64,65,66][55][56][57][58]. Furthermore, IDPs/IDRs exhibit different degrees of foldability, ranging from potentially foldable to not foldable at all [59,60,61,67][51][52][53][59].
IDPs/IDRs do not possess a precise equilibrium value of the atomic coordinates and backbone Ramachandran angles over time; as a result, they appear as “protein clouds” [68][60]. Despite being highly dynamic, the structures of such protein clouds are best described as a few low-energy conformations [69,70][61][62].
In most cases, upon interaction with specific binding partners, a disordered protein/segment undergoes a disorder to order conformational transition (termed as ‘induced folding’) [27[19][20][34][35][36][39][62][63][64][65][66][67],28,42,43,44,47,70,71,72,73,74,75],. Additionally, a disordered protein can bind to multiple partners to attain distinct conformations with each of them, which, in turn, enable it to interact with different targets [28,76,77][20][68][69]. Moreover, there is also a “conformational preference” for the structure attained by IDPs upon binding [78,79][70][71]. Post-translational modifications (PTMs) also enable IDPs/IDRs to attain diverse conformations, thus increasing the total repertoire of structures resulting from an IDP/IDR sequence [42,72,80,81,82,83,84][34][64][72][73][74][75][76]. Ensembles of the conformations resulting from a single sequence make it possible for IDPs to perform multiple apparently unrelated biological functions (termed as ‘moonlighting’) required for the maintenance of life [85,86,87,88,89][77][78][79][80][81].
2.4. Functional Classification of Intrinsically Disordered Proteins
Several classification schemes have been proposed over the years based on the functions performed by IDPs/IDRs [42,90][34][82]. Tompa et al. annotated IDPs/IDRs in six different functional categories depending on the presence/absence and the strength of the binding of the disordered proteins/regions to their ligands [29,91][21][83]. Later, this stratification was further extended to define eight functional classes of IDPs, namely entropic chains, modification sites, disordered chaperones, molecular effectors, molecular recognition assemblers, molecular recognition scavengers, metal sponges, and unknown, as shown in Figure 2 [92][84]. These functional subtypes can be present either alone or in combination within the same protein if the protein has several disordered regions [59,60,61][51][52][53]. In the following sections, different functional classes are described in some detail with relevant examples.
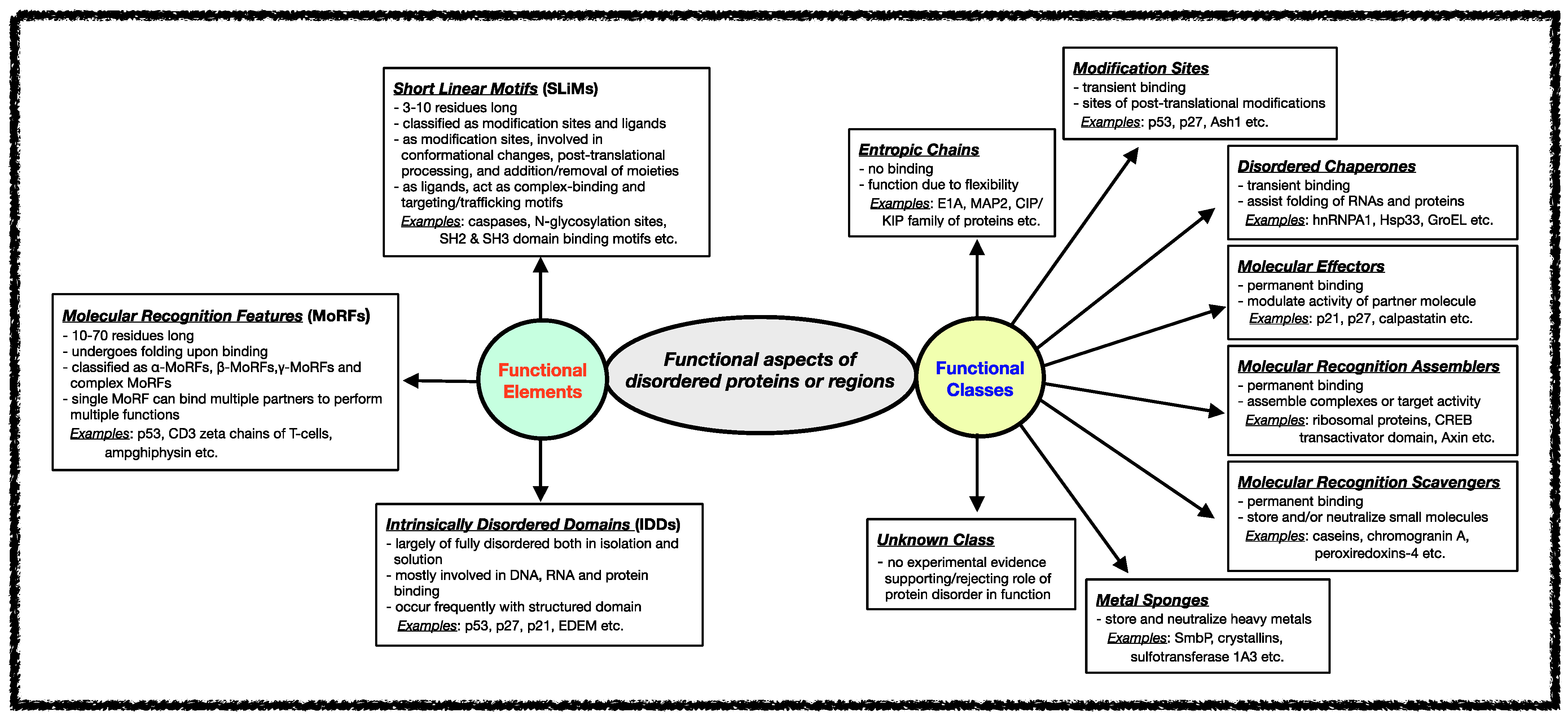
Figure 2. Functional aspects of IDPs/IDRs. Intrinsically disordered proteins/regions’ functional classes and elements are described here. The functional class scheme describes eight different categories into which IDPs/IDRs can be grouped based on their biological function. The different functional classes include Entropic chains, Modification sites, Disordered chaperones, Molecular effectors, Molecular recognition assemblers, Molecular recognition scavengers, Metal sponges, and Unknown. IDPs/IDRs’ functions are mediated mainly through three types of structural elements, namely Short Linear Motifs (SLiMs), Molecular Recognition Features (MORFs), and Intrinsically Disordered Domains (IDDS).
2.5. Functional Elements of Intrinsically Disordered Proteins
Various functional regions in intrinsically disordered regions have been revealed when studying different classes of functions carried out by IDRs. In general, the functional modules within IDRs have been classified into three categories: (i) Short Linear Motifs (SLiMs), (ii) Molecular Recognition Features (MoRFs), and (iii) Intrinsically Disordered Domains (IDDs) [78,160,161,162,163,164,165][70][85][86][87][88][89][90]. The classification and features of each of these functional modules are shown in Figure 2.
3. Experimental Approaches for Assessing Intrinsic Protein Disorder
Intrinsic protein disorder can be recognized and characterized by various direct and indirect (bio)physical methods. In contrast to direct techniques, which provide structural information about proteins, indirect approaches do not offer any structural details. Still, they suggest a behavior from which the disordered nature of the proteins can be inferred.3.1. Indirect Methods
Early understanding of the intrinsic structural disorders of proteins was based on a few simple techniques. In general, these indirect intrinsic disorder-identification approaches can quickly provide ample insight into the structural states of a protein or its segments. Because of the unusual amino acid composition and lack of a compact hydrophobic core, disordered proteins are evident during the purification process. Usually, the molecular mass (Mw) of IDPs estimated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) is higher by a factor of 1.2–1.8 in comparison with that measured by mass spectrometry [29][21]. Indeed, due to the enrichment of acidic residues and extension in solution, IDPs bind less to SDS and migrate more slowly on the gel in comparison with globular proteins [208][91]. The aberrant mobility of IDPs is also observed in size-exclusion chromatography (SEC) or gel-filtration (GF) experiments, as a result of which the apparent Mw of proteins with disordered regions is higher [45][37]. Furthermore, the flexible regions of proteins are known to have increased sensitivity to proteolytic degradation. IDPs, which are more affected than ordered proteins, based on limited in vitro proteolysis, exhibit high inherent flexibility [29,42,45,72][21][34][37][64]. Other peculiar biochemical behaviors of IDPs/IDRs include insensitivity to high temperatures and stability under acidic treatment. The resistance of IDPs/IDRs to boiling temperatures and acidic pH values has been ascribed to their lower contents of hydrophobic residues and enrichment of polar/charged residues, respectively [209,210,211][92][93][94]. Neutralizing acidic groups at lower pH levels reduces the net charge on IDPs/IDRs, leading to their increased solubility and a more compact structural state [45][37]. In contrast to IDPs/IDRs, the aggregation/precipitation of globular/ordered proteins occur at elevated temperatures and under low-pH conditions. While high-temperature conditions expose the hydrophobic core of ordered proteins, acidic conditions cause protonation of their negatively charged side chains, leading to charge imbalances, followed by the disruption of salt bridges and aggregate formation [29][21].3.2. Direct Methods
Several techniques provide both steady-state and dynamic structural information on IDPs/IDRs at the residue level. These methods capitalize on the significantly distinct conformational behavior of IDPs compared with that of globular proteins [29][21]. Some of the most commonly used direct methods are as follows:3.2.1. X-ray Crystallography
The diffraction intensity and X-ray pattern scattered by electrons in the protein structure are used to construct a three-dimensional (3-D) model of electron density, which, in turn, is used to deduce the atomic nuclei positions in the protein molecule [29][21]. Disordered regions in X-ray structures appear as missing regions [28][20]. This method can provide protein structure resolution down to 1Å. Still, additional experimental support is required to be certain about the structural disorder, as missing electron density regions can also result from technical failures in crystallography [212][95].3.2.2. Circular Dichroism (CD)
Circular dichroism (CD) is an absorption spectroscopy-based approach that relies on measuring the difference in the absorption spectra of right-handed and left-handed circularly polarized light. Optically active chiral molecules preferentially absorb either right-handed or left-handed circularly polarized light. Near-UV (250–350 nm) and far-UV (190–230 nm) CD signals are generally used to determine different aspects of the structure of proteins in solutions. The near-UV CD spectrum represents the tertiary structure around aromatic residues Phe, Tyr, and Trp [213,214][96][97]. While intense and detailed spectra characterize ordered proteins, those of IDPs are of low intensity and low complexity. The far-UV CD spectra of the secondary structural elements of proteins are quite distinct; therefore, they are used to determine the proportion of ⍺-helix, β-sheet, turn, PPII helix, and coil conformations in proteins [215][98]. If the far-UV CD spectrum is indicative of predominantly coil conformations, it indicates the disordered nature of the protein. In the case of proteins having both disordered and ordered regions, the CD does not provide clear information, as it lacks residue-specific details [28][20].3.2.3. Nuclear Magnetic Resonance (NMR)
NMR is the most common quantitative technique used for studying IDPs. The spinning ability of the charged atomic nuclei forms the basis of the 3-D structure determination of proteins in solutions using NMR. The directions of these spins are random, but the application of the external magnetic field can align these nuclei in directions either parallel or antiparallel to the applied magnetic field. These two states of nuclei have different energy levels, a low-energy state and a high-energy state. The low-energy state attains a high-energy state upon irradiation with electromagnetic radiation, and free inductive decay (FID) is obtained as the nuclei undergo relaxation. Fourier transformation of FID results in a NMR spectrum with peaks from different types of nuclei in the molecule, which, in turn, is used to characterize the local covalent and spatial arrangement of atoms [29,47,216,217][21][39][99][100]. In a protein, each nucleus of the individual residues experiences a different magnetic field depending on its microenvironment (referred to as the ‘shielding effect’ or ‘chemical shift’). The chemical shift of the peptide backbone (1H⍺, 13CO, 13C⍺, and 13Cβ) can be used to determine the secondary structure type of the given peptide segment [218,219][101][102]. Amino acids in ordered proteins are packed in different kinds of chemical environments, as a result of which their NMR spectrum resembles a combination of spectra of various secondary structure elements. In contrast, the NMR spectra of disordered proteins with extensive conformational averaging appear as a summation of the random coil spectra of residues of proteins [216,218][99][101]. In addition to the fine structural details, NMR also provides specific information at the residue level [28][20].3.2.4. Small-Angle X-ray Scattering (SAXS)
The SAXS technique can quickly define the structural characteristics of proteins of sizes ranging from a few kilo-Daltons to several giga-Daltons under various experimental setups [220,221,222,223][103][104][105][106]. Briefly, this method involves exposing samples placed in quartz capillary tubes to a collimated monochromatic X-ray beam source and capturing scattered photons with a detector [224][107]. Comparative analysis of the electron density distributions of the protein sample and pure solvent/buffer is then conducted to determine various parameters of the proteins in the solution, such as the molecular mass, volume, radius of gyration, folding state, etc. [220][103]. Moreover, SAXS data can also be used to define protein flexibility and the intrinsically disordered state of proteins in solutions [223,225][106][108]. The scattering profiles of the proteins obtained from SAXS experiments are most commonly represented as Kratky plots (s2I(s) as a function of s, where s and I represent the momentum transfer function and scattering intensity), which are used to obtain structural insights into the protein. In contrast to globular proteins’ bell-shaped Kratky’s plot with well-defined maxima, disordered protein-specific Kratky plots exhibit a plateau for a given range of the momentum transfer function (s), followed by a monotonic increase [226,227][109][110]. Additionally, the experimentally determined radius of gyration (Rg) of IDPs from the SAXS curve can be directly compared with the theoretical or experimental Rg values of a globular and random coil for a given number of residues. The Rg values of IDPs lie between those of highly compact globular proteins (lowest Rg values) and completely disordered/unfolded proteins represented by random coils (highest Rg values) [228][111]. Altogether, this method offers fast structural characterization of proteins in solutions with a relatively easy sample preparation protocol and can capture data under near-native conditions [229,230][112][113]. As the sensitivity of SAXS depends on the particle size, prior removal of the macromolecular aggregates during sample preparation using a method such as sedimentation or size-exclusion chromatography is suggested [231][114]. Finally, it is worth mentioning that SAXS-based studies of IDPs can use valuable a priori complementary information from several other experimental and in silico protein structure determination methods. For instance, X-ray crystallography depicts the structured regions of a protein, while SAXS defines the protein segments with missing electron density [232][115]. Similarly, NMR provides information about different domains/complex sub-units during analyses of bimolecular complexes and multi-domain proteins, and SAXS defines their relative inter-domain positions [233][116]. Furthermore, other complementary techniques, such as CD, spectroscopy, chromatography, etc., and SAXS, can be used for the biophysical characterization of IDPs [234][117]. A low-resolution protein structure defined through the ab initio modeling of SAXS data alone can be further refined using inputs from protein structure prediction tools, such as I-TASSER, CORAL, etc. [235,236][118][119]. Recently, various protein structure determination/prediction techniques and SAXS have been used to characterize partially disordered mycobacterial ESX-secretion-associated protein K (EspK) [237][120].3.2.5. Cryo-Electron Microscopy (Cryo-EM)
In the last five years, the research area involving the structural characterization of proteins and other biological entities has been revolutionized by the development of cryo-electron-microscopy-based techniques [238,239,240,241][121][122][123][124]. These methods overcome the limitations of primary methods, i.e., X-ray crystallography and NMR, and allow the structural characterization of relatively large, structurally heterogeneous, flexible, and dynamic assemblies at sub-nanometer atomic resolution (below 4 Å) [241,242,243][124][125][126]. Typically, a cryo-EM workflow contains three main steps: (a) vitrification (rapid cooling without ice crystal formation) of specimens in an aqueous solution, (b) image acquisition at a low electron dose using electron microscopy, and (c) 3D model reconstruction and validation. Single-particle analysis (SPA) and sub-tomogram averaging (STA) models are most commonly used for the structural annotation of proteins [244][127]. However, while the globular/ordered/structured regions of proteins can be structurally resolved using cryo-EM, the predicted intrinsically disordered regions in the proximity of flexible regions escape structural assignment [245][128]. Therefore, similar to x-ray crystallography, a high degree of intrinsic disorder restricts the implementation of cryo-EM techniques. Alternatively, the structure and dynamics of IDPs/IDRs can be investigated by complementing higher-resolution NMR studies of IDRs with the modeling capabilities of cryo-EM [246,247][129][130]. In conclusion, 3D cryo-EM maps in conjunction with high-resolution data from NMR can model IDPs under physiologically relevant conditions and provide insights into their functional behavior [243][126].4. Computational Tools for Disorder Prediction
The biased amino acid compositions and peculiar sequence characteristics of IDPs/IDRs have encouraged the development of various reliable computational tools for studying intrinsic protein disorders. As a result, disorder predictors have been grouped into three distinct classes based on the underlying concepts.4.1. Propensity-Based Predictors
In principle, a disorder predictor is classified as propensity-based if it depends on some essential physical or chemical characteristics of residues or on prior knowledge of the biological background of intrinsic protein disorder. Disorder-predicting tools, such as FoldIndex, NORSp, GlobPlot, CH plot, and PreLink belong to this category [45,245,246,247,248,249,250,251][37][128][129][130][131][132][133][134].4.2. Machine Learning Algorithms (MLAs) Based Predictors
This class of advanced predictors relies on algorithms trained on data sets of experimentally characterized disordered regions and can differentiate disorder and order encoding sequences [29][21]. Currently, the experimentally characterized disordered proteins are publicly available on three databases: MobiDB (http://mobidb.bio.unipd.it/; accessed on 7 November 2022), IDEAL (https://ngdc.cncb.ac.cn/databasecommons/database/id/198; accessed on 7 November 2022), and DisProt (http://www.disprot.org/; accessed on 7 November 2022) [92,252,253][84][135][136]. PONDR, Spritz, DisEMBL, RONN, and DISOPRED are a few predictors that fit into this category [254,255,256,257,258,259][137][138][139][140][141][142]. Recently, the field of protein structure prediction has been revolutionized by the development of the deep learning-based method AlphaFold [260][143]. This software generates a per-residue confidence score (pLDDT) based on the protein’s amino acid sequence. The most recent version of this tool, i.e., Alphafold2, has been reported to achieve protein structure prediction accuracy competitive with that of experimental determination [261,262,263][144][145][146]. However, this program gives a low confidence score (pLDDT < 50) for intrinsically unstructured or disordered proteins/regions, and the inconclusive predicted structure resembles a ribbon. In addition, this method does not anticipate the relative likelihood of diverse IDP conformations and the folding pathways followed by IDPs/IDRs attaining an ordered structure upon interaction with other biomolecules [70,264][62][147]. At present, the AlphaFold Protein Structure Database is considered as the most complete and precise representation of the human proteome [265,266][148][149].4.3. Inter-Residue Contact-Based Predictors
Predictors based on the idea that IDPs/IDRs are disordered because they cannot make enough inter-residue contacts required to compensate for the loss of configurational entropy during folding are grouped together as inter-residue contact-based predictors. The above conclusions may be derived by either simple statistics involving contact numbers or through sophisticated techniques of determining the total stabilization energy of a protein. Computational tools, such as IUPred, FoldUnfold, and Ucon belong to this class [267,268,269,270][150][151][152][153]. At present, there is no “best” disorder prediction computational tool. Therefore, to avoid the limitations of a given tool, prediction results from different disorder predictors relying on distinct principles should be combined to provide a consensus prediction, as implemented by meta-predictors (for example, PONDR-FIT) [271][154]. Alternatively, publicly available meta-servers (for example, MeDor and metaPRDOS can also be used for quick and simultaneous analysis of protein disorder using multiple predictors [272,273][155][156]. In several recent articles, extensive comparisons of various computational disorder prediction methods’ performance and comprehensive online resources useful for studying IDPs/IDRs were provided [274,275,276][157][158][159].5. Evolution of IDPs/IDRs
The evolution of proteins involves changes in the form of insertions, deletions, or substitutions in their amino acid sequences. Over time, such changes can accumulate in the proteins, giving rise to taxonomic classes having substantial differences in their amino acid compositions [277][160]. In general, the structure and function of proteins are well conserved, but several exceptions exist. Several previous studies suggested that, even if the protein sequence diverges extensively, the protein function is well-conserved [278][161]. Hence, proteins are generally considered as the ‘chemical fingerprints’ of evolutionary history, as they manifest the underlying genetic changes as amino acid sequences. The evolution of intrinsic disorder exhibits a wavy pattern in which highly disordered primordial proteins with predominantly RNA-chaperone-like activities were slowly replaced with highly structured proteins [118,279][162][163]. Later, because of its peculiar features regarding the regulation of complex cellular processes, protein disorder was reinvented at various succeeding evolutionary stages, resulting in the creation of more complex organisms from the last universal ancestor [280,281][164][165]. Several mechanisms, such as de novo generation, horizontal gene transfer, and lateral gene transfer, can give rise to genes that encode IDPs [57,282][49][166]. Approximately 14% of Pfam domains, predicted to be mostly disordered and shared by many protein families, appear to have originated from domain duplications and module exchange between genes [196][167]. The high frequency of occurrence of tandemly repeated sequences in IDPs/IDRs suggests that the expansion of internal repeat regions (microsatellite and minisatellite coding regions) is another possible way by which the IDPs encoding genes arose [283,284][168][169]. Looking at the exceptional functional variability conferred to IDPs/IDRs due to the genetic instability of repetitive elements, the mechanism of the extension of repeat elements appears as the frequent method of disorder spread during evolution and rapid genomic changes in adaptation [36,285,286][28][170][171]. Furthermore, these IDPs/IDRs can also act as hot spots for mutations, leading to the loss of different functional modalities and thus resulting in various types of diseases, including cancer [287,288,289][172][173][174]. Seera and Nagarajaram have recently shown that the disease-causing missense mutations within IDRs reduce the overall conformation heterogeneity of the IDRs as compared to their wild type counterparts, and the few ‘locked’ dominant conformations presumably limit their interaction with the cognate partners [290][175]. Recent studies have shown that disordered protein segments are encoded by GC-enriched gene regions, which, in turn, directly correspond to the disorderedness of the encoded proteins [291,292][176][177]. This GC enrichment is due to the prevalence of amino acids coded by GC-rich codons (G, A, R, and P) in the disordered regions of proteins [291][176]. At the residue level, a relatively higher rate of evolutionary changes in the disordered regions of proteins was observed compared with that in the ordered/globular domains, as there were no structural constraints to maintaining a 3-D structure [293][178]. However, in certain cases, structured domains and disordered regions of proteins have been observed to co-evolve at higher rates [294,295][179][180]. Despite these rapid changes, the biological functions of the structured domains and disordered regions are always conserved [296][181]. Hence, a deeper understanding of the conformation ensemble–function relationship will help to decipher the evolutionary trajectory of IDPs. Based on the conservation of sequences coding for protein disorder, disordered residues have been classified as constrained (both sequence and disorder are conserved) or flexible (only protein disorder is conserved). Together, constrained and flexible disorder residues are known as conserved disorder. On the other hand, if neither disorder nor the residues encoding it are conserved, such a disorder class is known as non-conserved disorder [297][182]. This integrated structural and evolutionary approach has recently been used to define the determinants of the functional adaptability of the neutrophin family of proteins involved in neuronal development [298][183]. Considering that the disordered regions in proteins have a distinct amino acid composition and evolutionary rate as compared with that of ordered regions, the substitution frequencies of residues in the disordered regions must also be distinct from those found in ordered regions. Thus, identifying the evolutionary and functional features of IDPs/IDRs has become a computational challenge, as most of the sequence analysis tools and parameter optimization procedures are aimed at ordered/structured regions of proteins. Recently, methods evaluating disordered proteins’ molecular features and sequence composition in a position-specific manner have been developed. These advancements have allowed researchers to pursue alignment-based evolutionary studies on IDPs/IDRs without aligning the residues discretely [299,300,301][184][185][186].6. IDPs/IDRs in Diseases
Like structured proteins, the expression, localization, and interactions of intrinsically disordered proteins (IDPs) are also highly coordinated and regulated. Multiple checkpoints at various stages of the expression of IDPs-specific genes (from transcript synthesis to protein degradation) ensure the availability of IDPs in appropriate quantities and for the desired duration, preventing any ectopic interactions [302][187]. Several studies have shown the role of IDPs/IDRs in different human disorders, including diabetes, cancer, amyloidosis, neurodegenerative, and cardiovascular diseases [303,304][188][189]. Some well-studied examples of IDPs associated with human disease are p53, Mdm2, PTEN, c-Myc, AF4, BRCA1, EWS, Bcl-2, c-Fos, HPV oncoproteins, etc. [303,305,306,307][188][190][191][192]. Moreover, the deposition of ⍺-synuclein, tau, and amyloid-β proteins leads to Alzheimer’s disease, the accumulation of ⍺-synuclein results in Parkinson’s disease, and aggregates of PrPSC cause prion diseases. The expansion of CAG triplet repeats in disease genes, which introduces disorder, results in the family of polyQ diseases, such as Kennedy’s disease, Huntington’s disease, etc. [308,309,310,311,312,313,314][193][194][195][196][197][198][199]. In the last two decades, the role of IDPs in human diseases has been actively studied, giving rise to new mechanistic findings that have led to the formation of the D2 concept (‘Disorder to Disorders’) [303][188]. Several comprehensive reviews and thematic series articles have been published covering the significance of IDPs in diseases [315,316,317][200][201][202]. For instance, Coskuner and Uversky described various hypotheses proposed to explain the molecular mechanisms of the pathogenesis of Alzheimer’s and Parkinson’s diseases and suggested the need for the development of new techniques through the integration of quantum and statistical mechanics, thermodynamics, bioinformatics, and machine learning approaches, which, in turn, may lead to the development of new experimental approaches [318,319,320,321,322,323,324,325][203][204][205][206][207][208][209][210]. However, at present, there are several limitations and challenges associated with in silico studies of IDP-associated neurodegenerative disorders [326,327][211][212]. Another study found that an NADH-stabilized 26S proteasomal complex could degrade IDPs efficiently. Therefore, the accumulation of disease-causing disordered proteins, such as tau, c-Fos, p53, etc., can be prevented by the selective degradation of IDPs in an ATP-independent manner [328][213]. Moreover, the analysis of components of the ATP-dependent ubiquitin-proteasome degradation system (UPS) revealed the importance of the disorder content and MoRFs of the complex in neurodegenerative disorders and cancers [329][214]. However, identifying key mutations, PTM sites, and functional motifs in the disordered regions, exploring the evolutionary history of IDPs involved in diseases, understanding the cooperative functioning of ordered and disordered domains, and dissecting the IDPs’ interactome are some of the many active research areas involving IDPs/IDRs and diseases [288,330,331,332,333,334][173][215][216][217][218][219].7. IDPs/IDRs as Drug Targets
With increasing evidence of their involvement in molecular functions complementing globular domains, essential biological processes, protein–nucleic acid interactions, protein–protein interactions, and diseases, IDRs/IDPs have emerged as one of the prime targets for drug discovery or repurposing [142,335,336,337,338,339][220][221][222][223][224][225]. However, IDP characteristics, such as a lack of a sTable 3D structure, very high flexibility, conformational ensembles, susceptibility to proteolytic cleavage, protein aggregation, etc., limit the application of the most-established experimental assays and computational methods that would otherwise work for ordered/globular proteins [340,341,342,343,344][226][227][228][229][230]. Therefore, IDP-specific drug screening/development is mainly a tradeoff between binding affinity/specificity and the alternation in the functioning of disordered proteins with other features, such as solubility, crowding, efflux, metabolism, etc., a potentially relevant role [345][231]. Broadly, disordered proteins/regions have been used in drug development procedures by targeting their conformational changes, interactions, and self-aggregating behavior [346][232]. For example, the inhibitor 10058F4 of Myc proto-oncogene protein (MYC) binds to MYC and prevents conformational disorder-to-order transition, which, in turn, blocks MYC-MAX complex-driven tumorigenesis [33,347,348,349,350][25][233][234][235][236]. Similarly, Methyl-CpG-binding domain protein 2 (MBD2) inhibitors restrict the folding of MBD2 upon binding to its partner p66α. This MBD2-p66α is known to regulate the Mi-2/NuRD chromatin remodeling complex involved in promoting metastasis in various cancer cells through epithelial–mesenchymal transition (EMT) [351,352][237][238]. In contrast to ordered proteins, the protein–protein interactions involving IDPs offer uneven, shorter, compact, and more mimicable surfaces for the tighter binding of small drug molecules [353,354,355][239][240][241]. In recent times, potential drug molecules have been designed to target either the disordered segment or the binding region of the interacting molecule. For instance, nutlins binding to Mdm2 prevent the interaction of Mdm2 with the disordered regions of p53, which activates the p53 pathway, leading to apoptosis, cell-cycle arrest, and the inhibition of the uncontrolled cell growth of human tumor xenografts [356][242]. Additionally, an FDA-approved compound, trifluoperazine dihydrochloride, was found to bind to a disordered region of multifunctional protein nuclear protein 1 (NUPR1) and arrest pancreatic ductal adenocarcinoma (PDAC) development [357][243]. Moreover, the disordered proteins from pathogens can also be targeted to interrupt their interaction with host proteins, which they utilize for their survival and pathogenesis [358][244]. In a recent review, Santofimia et al. comprehensively described targeting IDPs in various protein–protein and protein–nucleic acid interactions involved in cancer [359][245]. Furthermore, compounds, such as curcumin, rosmarinic acid, ferulic acid, and safranal, have also been reported to prevent the aggregation of α-synuclein protein by binding to monomers, thus inhibiting the polymerization of these proteins, which results in various neuronal malignancies [360,361][246][247]. In summary, deciphering the sequence–ensemble–function relationship of IDPs/IDRs and the development of efficient computational modeling approaches will help to unravel the enormous potential of disordered proteins as drug targets.References
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Structural biology. Versatility from protein disorder. Science 2012, 337, 1460–1461.
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631.
- Luo, Y.; Ye, S.; Li, X.; Lu, W. Emerging Structure-Function Paradigm of Endocrine FGFs in Metabolic Diseases. Trends Pharmacol. Sci. 2019, 40, 142–153.
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783.
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17.
- Mak, H.; Thurston, T.L.M. Interesting Biochemistries in the Structure and Function of Bacterial Effectors. Front. Cell Infect. Microbiol. 2021, 11, 608860.
- Pinet, L.; Assrir, N.; van Heijenoort, C. Expanding the Disorder-Function Paradigm in the C-Terminal Tails of Erbbs. Biomolecules 2021, 11, 1690.
- Ghag, G.; Holler, C.J.; Taylor, G.; Kukar, T.L.; Uversky, V.N.; Rangachari, V. Disulfide bonds and disorder in granulin-3: An unusual handshake between structural stability and plasticity. Protein Sci. 2017, 9, 1759–1772.
- Davey, N.E. The functional importance of structure in unstructured protein regions. Curr. Opin. Struct. Biol. 2019, 56, 155–163.
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. On the roles of intrinsically disordered proteins and regions in cell communication and signaling. Cell Commun. Signal. 2021, 19, 88.
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157.
- Bickers, S.C.; Sayewich, J.S.; Kanelis, V. Intrinsically disordered regions regulate the activities of ATP binding cassette transporters. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183202.
- Brodsky, S.; Jana, T.; Mittelman, K.; Chapal, M.; Kumar, D.K.; Carmi, M.; Barkai, N. Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 2020, 79, 459–471.
- Jamecna, D.; Antonny, B. Intrinsically disordered protein regions at membrane contact sites. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159020.
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174.
- Protter, D.S.W.; Rao, B.S.; Van Treeck, B.; Lin, Y.; Mizoue, L.; Rosen, M.K.; Parker, R. Intrinsically Disordered Regions Can Contribute Promiscuous Interactions to RNP Granule Assembly. Cell Rep. 2018, 22, 1401–1412.
- Macossay-Castillo, M.; Marvelli, G.; Guharoy, M.; Jain, A.; Kihara, D.; Tompa, P.; Wodak, S.J. The Balancing Act of Intrinsically Disordered Proteins: Enabling Functional Diversity while Minimizing Promiscuity. J. Mol. Biol. 2019, 431, 1650–1670.
- Chakrabarti, P.; Chakravarty, D. Intrinsically disordered proteins/regions and insight into their biomolecular interactions. Biophysical Chemistry 2022, 283, 106769.
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331.
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59.
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533.
- Uversky, V.; Gillespie, J.; Fink, A. Why Are “Natively Unfolded” Proteins Unstructured Under Physiologic Conditions? Proteins Struct. Funct. Genet. 2000, 41, 415–427.
- Wallin, S. Intrinsically disordered proteins: Structural and functional dynamics. Res. Rep. Biol. 2017, 8, 7–16.
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, G.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484.
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1231–1264.
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103.
- Uversky VN The mysterious unfoldome: Structureless, under appreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068.
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645.
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C. Intrinsic Protein Disorder in Complete Genomes. Genome Inform. 2000, 11, 161–171.
- Hu, G.; Wang, K.; Song, J.; Uversky, V.N.; Kurgan, L. Taxonomic Landscape of the Dark Proteomes: Whole-Proteome Scale Interplay Between Structural Darkness, Intrinsic Disorder, and Crystallization Propensity. Proteomics 2018, 18, e1800243.
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Longhi, S.; Zhang, K.Y.J.; Bhatia, S. Comprehensive Intrinsic Disorder Analysis of 6108 Viral Proteomes: From the Extent of Intrinsic Disorder Penetrance to Functional Annotation of Disordered Viral Proteins. J. Proteome Res. 2021, 20, 2704–2713.
- Monzon, A.M.; Necci, M.; Quaglia, F.; Walsh, I.; Zanotti, G.; Piovesan, D.; Tosatto, S.C.E. Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. Int. J. Mol. Sci. 2020, 21, 4496.
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent Structural Disorder in E. coli and S. cerevisiae Proteomes. J. Proteome Res. 2006, 5, 1996–2000.
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic Disorder and Protein Function. Biochemistry 2002, 41, 6573–6582.
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148.
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208.
- Uversky, V. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12.
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756.
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60.
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic Disorder and Functional Proteomics. Biophys. J. 2007, 92, 1439–1456.
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.; Brown, C.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Genet. 2001, 42, 38–48.
- Williams, R.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 6, 89–100.
- Holt, C.; Raynes, J.K.; Carver, J.A. Sequence characteristics responsible for protein-protein interactions in the intrinsically disordered regions of caseins, amelogenins, and small heat-shock proteins. Biopolymers 2019, 110, e23319.
- Kastano, K.; Mier, P.; Dosztanyi, Z.; Promponas, V.J.; Andrade-Navarro, M.A. Functional Tuning of Intrinsically Disordered Regions in Human Proteins by Composition Bias. Biomolecules 2022, 12, 1486.
- Trivedi, R.; Nagarajaram, H.A. Amino acid substitution scoring matrices specific to intrinsically disordered regions in proteins. Sci Rep. 2019, 9, 16380.
- Trivedi, R.; Nagarajaram, H.A. Substitution scoring matrices for proteins—An overview. Protein Sci. 2020, 29, 2150–2163.
- Jarnot, P.; Ziemska-Legiecka, J.; Grynberg, M.; Gruca, A. Insights from analyses of low complexity regions with canonical methods for protein sequence comparison. Brief Bioinform. 2022, 23, bbac299.
- McFadden, W.M.; Yanowitz, J.L. idpr: A package for profiling and analyzing Intrinsically Disordered Proteins in R. PLoS ONE 2022, 17, e0266929.
- Tompa, P. Structure and Function of Intrinsically Disordered Proteins; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Abingdon, UK, 2010.
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384.
- Uversky, V. Intrinsic Disorder-based Protein Interactions and their Modulators. Curr. Pharm. Des. 2013, 19, 4191–4213.
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901.
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 932–951.
- Morffy, N.; Strader, L.C. Structural Aspects of Auxin Signaling. Cold Spring Harb. Perspect. Biol. 2022, 14, a039883.
- Crick, S.L.; Jayaraman, M.; Frieden, C.; Wetzel, R.; Pappu, R.V. Fluorescence correlation spectroscopy shows that monomeric poly- glutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA 2006, 103, 16764–16769.
- Walters, R.H.; Murphy, R.M. Examining Polyglutamine Peptide Length: A Connection between Collapsed Conformations and Increased Aggregation. J. Mol. Biol. 2009, 393, 978–992.
- Mukhopadhyay, S.; Krishnan, R.; Lemke, E.A.; Lindquist, S.; Deniz, A.A. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. USA 2007, 104, 2649–2654.
- Wang, X.; Vitalis, A.; Wyczalkowski, M.A.; Pappu, R.V. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins Struct. Funct. Bioinform. 2006, 63, 297–311.
- Mukhopadhyay, S. The Dynamism of Intrinsically Disordered Proteins: Binding-Induced Folding, Amyloid Formation, and Phase Separation. J. Phys. Chem. B 2020, 124, 11541–11560.
- Dunker, A.K.; Uversky, V.N. Drugs for ‘protein clouds’: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788.
- Choy, W.Y.; Forman-Kay, J.D. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J. Mol. Biol. 2001, 308, 1011–1032.
- Strodel, B. Energy Landscapes of Protein Aggregation and Conformation Switching in Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167182.
- Dunker, A.K.; Obradovic, Z. The protein trinity—Linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806.
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and Functions of Usefully Disordered Proteins. Adv. Protein Chem. 2002, 62, 25–49.
- Toto, A.; Malagrino, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated folding of intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 6586–6593.
- Malagrino, F.; Visconti, L.; Pagano, L.; Toto, A.; Troilo, F.; Gianni, S. Understanding the Binding Induced Folding of Intrinsically Disordered Proteins by Protein Engineering: Caveats and Pitfalls. Int. J. Mol. Sci. 2020, 21, 3484.
- Malagrino, F.; Diop, A.; Pagano, L.; Nardella, C.; Toto, A.; Gianni, S. Unveiling induced folding of intrinsically disordered proteins—Protein engineering, frustration and emerging themes. Curr. Opin. Struct. Biol. 2022, 72, 153–160.
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p2lWa1CiPl/Sdil in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509.
- Uversky, V.N. Analyzing IDPs in Interactomes. Methods Mol Biol. 2020, 2141, 895–945.
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed Structural Elements Feature in Partner Recognition by Intrinsically Unstructured Proteins. J. Mol. Biol. 2004, 338, 1015–1026.
- Choi, U.B.; Sanabria, H.; Smirnova, T.; Bowen, M.E.; Weninger, K.R. Spontaneous Switching among Conformational Ensembles in Intrinsically Disordered Proteins. Biomolecules 2019, 9, 114.
- Follis, A.V.; Llambi, F.; Kalkavan, H.; Yao, Y.; Phillips, A.H.; Park, C.G.; Marassi, F.M.; Green, D.R.; Kriwacki, R.W. Regulation of apoptosis by an intrinsically disordered region of Bcl-xL. Nat. Chem. Biol. 2018, 14, 458–465.
- Miao, Y.; Tipakornsaowapak, T.; Zheng, L.; Mu, Y.; Lewellyn, E. Phospho-regulation of intrinsically disordered proteins for actin assembly and endocytosis. FEBS J. 2018, 285, 2762–2784.
- Csizmok, V.; Forman-Kay, J.D. Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications. Curr. Opin. Struct. Biol. 2018, 48, 58–67.
- Bludau, I.; Willems, S.; Zeng, W.F.; Strauss, M.T.; Hansen, F.M.; Tanzer, M.C.; Karayel, O.; Schulman, B.A.; Mann, M. The structural context of posttranslational modifications at a proteome-wide scale. PLoS Biol. 2022, 20, e3001636.
- Ye, H.; Han, Y.; Li, P.; Su, Z.; Huang, Y. The Role of Post-Translational Modifications on the Structure and Function of Tau Protein. J. Mol. Neurosci. 2022, 72, 1557–1571.
- Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11.
- Jeffery, C.J. Molecular mechanisms for multitasking: Recent crystal structures of moonlighting proteins. Curr. Opin. Struct. Biol. 2004, 14, 663–668.
- Meng, F.; Kurgan, L. High-throughput prediction of disordered moonlighting regions in protein sequences. Proteins 2018, 86, 1097–1110.
- Balcerak, A.; Trebinska-Stryjewska, A.; Konopinski, R.; Wakula, M.; Grzybowska, E.A. RNA-protein interactions: Disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 2019, 9, 190096.
- Rafiee, M.R.; Zagalak, J.A.; Sidorov, S.; Steinhauser, S.; Davey, K.; Ule, J.; Luscombe, N.M. Chromatin-contact atlas reveals disorder-mediated protein interactions and moonlighting chromatin-associated RBPs. Nucleic Acids Res. 2021, 49, 13092–13107.
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103.
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354.
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793.
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. BioSyst. 2012, 8, 268–281.
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956.
- Lee, S.; Kim, D.; Han, J.; Cha, E.; Lim, J.; Cho, Y.; Lee, C.; Han, K. Understanding Pre-Structured Motifs (PreSMos) in Intrinsically Unfolded Proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54.
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059.
- Oldfield, C.; Cheng, Y.; Cortese, M.; Romero, P.; Uversky, V.; Dunker, A.K. Coupled Folding and Binding with R-Helix-Forming Molecular Recognition. Biochemistry 2005, 44, 12454–12470.
- Hinds, M.G.; Smits, C.; Fredericks-Short, R.; Risk, J.M.; Bailey, M.; Huang, D.C.; Day, C.L. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007, 14, 128–136.
- Blocquel, D.; Habchi, J.; Gruet, A.; Blangy, S.; Longhi, S. Compaction and binding properties of the intrinsically disordered C-terminal domain of Henipavirus nucleoprotein as unveiled by deletion studies. Mol. BioSyst. 2012, 8, 392–410.
- Etoh, Y.; Simon, M.; Green, H. Involucrin acts as transglutaminase substrate at multiple sites. Biochem. Biophys. Res. Commun. 1986, 136, 51–60.
- Lynch, W.; Riseman, V.; Bretscher, A. Smooth Muscle CaldesmonIs an Extended Flexible Monomeric Protein in Solution That Can Readily Undergo Reversible Intra- and Intermolecular Sulfhydryl Cross-linking. J. Biol. Chem. 1987, 262, 7429–7437.
- Weinreb, P.; Zhen, W.; Poon, A.; Conway, K.; Lansbury, P.J. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715.
- Huber, R. Conformational flexibility in protein molecules. Nature 1979, 280, 538–539.
- Ohgushi, M.; Wada, A. ‘Molten-globule state’: A compact form of globular proteins with mobile side chains. FEBS Lett. 1983, 164, 21–24.
- Kuwajima, K. A Folding Model of a-Lactalbumin Deduced from the Three-state Denaturation Mechanism. J. Mol. Biol. 1977, 114, 241–258.
- Fasman, G.D. Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA, 1996.
- Dyson, H.J.; Wright, P.E. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 2002, 62, 311–340.
- Dyson, H.J.; Wright, P.E. Unfolded proteins and protein folding studied by NMR. Chem. Rev. 2004, 104, 3607–3622.
- Poulsen, F.M. A Brief Introduction to NMR Spectroscopy of Proteins. 2002. Available online: https://users.cs.duke.edu/~brd/Teaching/Bio/asmb/Papers/Intro-reviews/flemming.pdf (accessed on 7 November 2022).
- Cavalli, A.; Salvatella, X.; Dobson, C.M.; Vendruscolo, M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2007, 104, 9615–9620.
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577.
- Korasick, D.A.; Tanner, J.J. Determination of protein oligomeric structure from small-angle X-ray scattering. Protein Sci. 2018, 27, 814–824.
- Da Vela, S.; Svergun, D.I. Methods, development and applications of small-angle X-ray scattering to characterize biological macromolecules in solution. Curr. Res. Struct. Biol. 2020, 27, 164–170.
- Grawert, T.W.; Svergun, D.I. Structural Modeling Using Solution Small-Angle X-ray Scattering (SAXS). J. Mol. Biol. 2020, 432, 3078–3092.
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology--Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657.
- Czaplewski, C.; Gong, Z.; Lubecka, E.A.; Xue, K.; Tang, C.; Liwo, A. Recent Developments in Data-Assisted Modeling of Flexible Proteins. Front. Mol. Biosci. 2021, 8, 765562.
- Perez, J.; Vachette, P.; Russo, D.; Desmadril, M.; Durand, D. Heat-induced unfolding of neocarzinostatin, a small all-β protein investigated by small-angle X-ray scattering. J. Mol. Biol. 2001, 308, 721–743.
- Bernado, P.; Blackledge, M. A Self-Consistent Description of the Conformational Behavior of Chemically Denatured Proteins from NMR and Small Angle Scattering. Biophys. J. 2009, 97, 2839–2845.
- Bernado, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167.
- Jeffries, C.M.; Svergun, D.I. High-Throughput Studies of Protein Shapes and Interactions by Synchrotron Small-Angle X-ray Scattering. In Structural Proteomics; Methods in Molecular Biology; Owens, R., Ed.; Humana: New York, NY, USA, 2015; Volume 1261, pp. 277–301.
- Graewert, M.A.; Jeffries, C.M. Sample and Buffer Preparation for SAXS. Adv. Exp. Med. Biol. 2017, 1009, 11–30.
- David, G.; Perez, J. Combined sampler robot and high-performance liquid chromatography: A fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009, 42, 892–900.
- Schulte, T.; Lofling, J.; Mikaelsson, C.; Kikhney, A.; Hentrich, K.; Diamante, A.; Ebel, C.; Normark, S.; Svergun, D.; Birgitta, H.N.; et al. The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Open Biol. 2014, 41, 30090.
- Rodriguez, Z.P. Conjugation of NMR and SAXS for flexible and multidomain protein structure determination: From sample preparation to model refinement. Prog. Biophys. Mol. Biol. 2020, 150, 140–144.
- Gast, K.; Damaschun, H.; Eckert, K.; Schulze-Forster, K.; Maurer, H.R.; Muller-Frohne, M.; Zirwer, D.; Czarnecki, J.; Damaschun, G. Prothymosin alpha: A biologically active protein with random coil conformation. Biochemistry 1995, 34, 13211–13218.
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40.
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350.
- Gijsbers, A.; Sánchez-Puig, N.; Gao, Y.; Peters, P.J.; Ravelli, R.B.G.; Siliqi, D. Structural Analysis of the Partially Disordered Protein EspK from Mycobacterium Tuberculosis. Crystals 2021, 11, 18.
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27.
- Shoemaker, S.C.; Ando, N. X-rays in the cryo-electron microscopy era: Structural biology’s dynamic future. Bichemistry 2018, 57, 277–285.
- Hanske, J.; Sadian, Y.; Muller, C.W. The cryo-em resolution revolution and transcription complexes. Curr. Opin. Struct. Biol. 2018, 52, 8–15.
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit. Int. J. Mol. Sci. 2019, 20, 4186.
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880.
- Musselman, C.A.; Kutateladze, T.G. Characterization of functional disordered regions within chromatin-associated proteins. IScience 2021, 24, 102070.
- Bharat, T.A.M.; Russo, C.J.; Lowe, J.; Passmore, L.A.; Scheres, S.H.W. Advances in single-particle electron cryo microscopy structure determination applied to sub-tomogram averaging. Structure 2015, 23, 1743–1753.
- Yan, Z.; Bai, X.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.; Li, X.; Scheres, S.H.W.; et al. Structure of the rabbit ryanodine receptor ryr1 at near-atomic resolution. Nature 2015, 517, 50–55.
- Gibbs, E.B.; Cook, E.C.; Showalter, S.A. Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys. 2017, 628, 57–70.
- Geraets, J.A.; Pothula, K.R.; Schroder, G.F. Integrating cryo-EM and NMR data. Curr. Opin. Struc. Biol. 2020, 61, 173–181.
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex(C): A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438.
- Liu, J.; Rost, B. NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res. 2003, 31, 3833–3835.
- Linding, R.; Russell, R.; Neduva, V.; Gibson, T. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708.
- Coeytaux, K.; Poupon, A. Prediction of unfolded segments in a protein sequence based on amino acid composition. Bioinformatics 2005, 21, 1891–1900.
- Di Domenico, T.; Walsh, I.; Martin, A.J.; Tosatto, S.C. MobiDB: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012, 28, 2080–2081.
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically Disordered proteins with Extensive Annotations and Literature. Nucleic Acids Res. 2012, 40, D507–D511.
- Romero, P.; Obradovic, Z.; Dunker, A.K. Folding minimal sequences: The lower bound for sequence complexity of globular proteins. FEBS Lett. 1999, 462, 363–367.
- Vullo, A.; Bortolami, O.; Pollastri, G.; Tosatto, S.C. Spritz: A server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006, 34, W164–W168.
- Linding, R.; Jensen, L.; Diella, F.; Bork, P.; Gibson, T.; Russell, R. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459.
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376.
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139.
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Zweckstetter, M. NMR hawk-eyed view of AlphaFold2 structures. Protein Sci. 2021, 30, 2333–2337.
- AlQuraishi, M. Protein-structure prediction revolutionized. Nature 2021, 596, 487–488.
- He, X.H.; You, C.Z.; Jiang, H.L.; Jiang, Y.; Xu, H.E.; Cheng, X. AlphaFold2 versus experimental structures: Evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 2022.
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208.
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444.
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596.
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434.
- Dosztanyi, Z.; Csizmok VTompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839.
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. To be folded or to be unfolded? Protein Sci. 2008, 13, 2871–2877.
- Schlessinger, A.; Punta, M.; Rost, B. Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 2007, 23, 2376–2384.
- Xue BDunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010.
- Lieutaud, P.; Canard, B.; Longhi, S. MeDor: A metaserver for predicting protein disorder. BMC Genom. 2008, 9, S25.
- Ishida, T.; Kinoshita, K. Prediction of disordered regions in proteins based on the meta-approach. Bioinformatics 2008, 24, 1344–1348.
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090.
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief Bioinform. 2019, 20, 330–346.
- Kurgan, L. Resources for computational prediction of intrinsic disorder in proteins. Methods 2022, 204, 132–141.
- Jordan, I.; Kondrashov, F.; Adzhubei, I.A.; Wolf, Y.; Koonin, E.; Kondrashov, A.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638.
- Brooks, D.J.; Fresco, J.R. Increased Frequency of Cysteine, Tyrosine, and Phenylalanine Residues Since the Last Universal Ancestor. Mol. Cell. Proteom. 2002, 1, 125–131.
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004, 18, 1169–1175.
- Cristofari, G.; Darlix, J.L. The ubiquitous nature of RNA chaperone proteins. Prog. Nucl. Acid Res. Mol. Biol. 2002, 72, 223–268.
- Doolittle, W.F. Uprooting the tree of life. Sci. Am. 2000, 282, 90–95.
- Theobald, D.L. A formal test of the theory of universal common ancestry. Nature 2010, 465, 219–222.
- Van Oss, S.B.; Carvunis, A.R. De novo gene birth. PLoS Genet. 2019, 15, e1008160.
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. BioEssays 2009, 31, 328–335.
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. BioEssays 2003, 25, 847–855.
- Lise, S.; Jones, D. Sequence Patterns Associated with Disordered Regions in Proteins. Proteins Struct. Funct. Genet. 2005, 58, 144–150.
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446.
- Pancsa, R.; Tompa, P. Structural Disorder in Eukaryotes. PLoS ONE 2012, 7, e34687.
- Meyer, K.; Kirchner, M.; Uyar, B.; Cheng, J.Y.; Russo, G.; Hernandez-Miranda, L.R.; Szymborska, A.; Zauber, H.; Rudolph, I.M.; Willnow, T.E.; et al. Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 2018, 175, 239–253.
- Pajkos, M.; Zeke, A.; Dosztányi, Z. Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules 2020, 10, 1115.
- Meszaros, B.; Hajdu-Soltesz, B.; Zeke, A.; Dosztanyi, Z. Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules 2021, 11, 381.
- Seera, S.; Nagarajaram, H.A. Effect of Disease Causing Missense Mutations on Intrinsically Disordered Regions in Proteins. Protein Pept. Lett. 2022, 29, 254–267.
- Peng, Z.; Uversky, V.; Kurgan, L. Genes encoding intrinsic disorder in Eukaryota have high GC content. Intrinsically Disord. Proteins 2016, 4, e1262225.
- Basile, W.; Sachenkova, O.; Light, S.; Elofsson, A. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput. Biol. 2017, 13, e1005375.
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary Rate Heterogeneity in Proteins with Long Disordered Regions. J. Mol. Evol. 2002, 55, 104–110.
- Homma, K.; Anbo, H.; Noguchi, T.; Fukuchi, S. Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins. Int. J. Mol. Sci. 2018, 19, 3860.
- Pancsa, R.; Zsolyomi, F.; Tompa, P. Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings. Int. J. Mol. Sci. 2018, 19, 3315.
- Zarin, T.; Strome, B.; Nguyen Ba, A.N.; Alberti, S.; Forman-Kay, J.D.; Moses, A.M. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. eLife 2019, 8, e46883.
- Bellay, J.; Han, S.; Michaut, M.; Kim, T.; Costanzo, M.; Andrews, B.J.; Boone, C.; Bader, G.D.; Myers, C.L.; Kim, P.M. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011, 12, R14.
- Covaceuszach, S.; Peche, L.Y.; Konarev, P.V.; Lamba, D. A combined evolutionary and structural approach to disclose the primary structural determinants essential for proneurotrophins biological functions. Comput. Struct. Biotechnol. J. 2021, 19, 2891–2904.
- Millard, P.S.; Bugge, K.; Marabini, R.; Boomsma, W.; Burow, M.; Kragelund, B.B. IDDomainSpotter: Compositional bias reveals domains in long disordered protein regions-Insights from transcription factors. Protein Sci. 2020, 29, 169–183.
- Cohan, M.C.; Shinn, M.K.; Lalmansingh, J.M.; Pappu, R.V. Uncovering Non-random Binary Patterns Within Sequences of Intrinsically Disordered Proteins. J. Mol. Biol. 2022, 434, 167373.
- Lu, A.X.; Lu, A.X.; Pritisanac, I.; Zarin, T.; Forman-Kay, J.D.; Moses, A.M. Discovering molecular features of intrinsically disordered regions by using evolution for contrastive learning. PLoS Comput. Biol. 2022, 18, e1010238.
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight Regulation of Unstructured Proteins: From Transcript Synthesis to Protein Degradation. Science 2008, 322, 1365–1368.
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246.
- Langella, E.; Buonanno, M.; De Simone, G.; Monti, S.M. Intrinsically disordered features of carbonic anhydrase IX proteoglycan-like domain. Cell. Mol. Life Sci. 2021, 78, 2059–2067.
- Chang, B.S.; Minn, A.J.; Muchmore, S.W.; Fesik, S.W.; Thompson, C.B. Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J. 1997, 16, 968–977.
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.; Suk, J.; Kim, D.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K. Local Structural Elements in the Mostly Unstructured Transcriptional Activation Domain of Human p53. J. Biol. Chem. 2000, 275, 29426–29432.
- Garg, N.; Kumar, P.; Gadhave, K.; Giri, R. The dark proteome of cancer: Intrinsic disorderedness and functionality of HIF-1α along with its interacting proteins. Prog. Mol. Biol. Transl. Sci. 2019, 166, 371–403.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135.
- Lee, V.; Balin, B.; Otvos, L.J.; Trojanowski, J. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678.
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286.
- Prusiner, S. Shattuck lecture: Neurodegenerative diseases and prions. N. Engl. J. Med. 2001, 344, 1516–1526.
- Eftekharzadeh BHyman, B.T.; Wegmann, S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech. Ageing Dev. 2016, 156, 1–13.
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307.
- Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322.
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147.
- Monti, S.M.; De Simone, G.; Langella, E. The Amazing World of IDPs in Human Diseases. Biomolecules 2021, 11, 333.
- Monti, S.M.; De Simone, G.; Langella, E. The Amazing World of IDPs in Human Diseases II. Biomolecules 2022, 12, 369.
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818.
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161.
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr. Med. Chem. 2018, 25, 3141–3159.
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Coskuner, O.; Uversky, V.N. Intrinsically disordered proteins in various hypotheses on the pathogenesis of Alzheimer’s and Parkinson’s diseases. Prog. Mol. Biol. Transl. Sci. 2019, 166, 145–223.
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164.
- Coskuner-Weber, O.; Mirzanli, O.; Uversky, V.N. Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases. Biophys. Rev. 2022, 14, 679–707.
- Akbayrak, I.Y.; Caglayan, S.I.; Ozcan, Z.; Uversky, V.N.; Coskuner-Weber, O. Current Challenges and Limitations in the Studies of Intrinsically Disordered Proteins in Neurodegenerative Diseases by Computer Simulations. Curr. Alzheimer Res. 2020, 17, 805–818.
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model. Int. J. Mol. Sci. 2021, 19, 10587.
- Tsvetkov, P.; Myers, N.; Adler, J.; Shaul, Y. Degradation of Intrinsically Disordered Proteins by the NADH 26S Proteasome. Biomolecules 2020, 10, 1642.
- Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules 2020, 10, 796.
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Molecular Context-Dependent Effects Induced by Rett Syndrome-Associated Mutations in MeCP2. Biomolecules 2020, 10, 1533.
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Stabilization Effect of Intrinsically Disordered Regions on Multidomain Proteins: The Case of the Methyl-CpG Protein 2, MeCP2. Biomolecules 2021, 11, 1216.
- Neira, J.L.; Rizzuti, B.; Jimenez-Alesanco, A.; Palomino-Schätzlein, M.; Abian, O.; Velazquez-Campoy, A.; Iovanna, J.L. A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin. Biomolecule 2020, 10, 1313.
- Wong, E.T.C.; So, V.; Guron, M.; Kuechler, E.R.; Malhis, N.; Bui, J.M.; Gsponer, J. Protein-Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules 2020, 10, 1097.
- Raut, K.K.; Ponniah, K.; Pascal, S.M. Structural Analysis of the cl-Par-4 Tumor Suppressor as a Function of Ionic Environment. Biomolecules 2021, 11, 3386.
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J Mol. Biol. 2018, 430, 2309–2320.
- Wang, J.; Cao, Z.; Zhao, L.; Li, S. Novel strategies for drug discovery based on Intrinsically Disordered Proteins (IDPs). Int. J. Mol. Sci. 2011, 12, 3205–3219.
- Melo, A.M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. USA 2016, 113, 14336–14341.
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 2017, 18, 2761.
- Necci, M.; Piovesan, D. CAID Predictors, DisProt Curators, Tosatto SCE Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481.
- Necci, M.; Piovesan, D.; Tosatto, S.C. Large-scale analysis of intrinsic disorder flavors and associated functions in the protein sequence universe. Protein Sci. 2016, 25, 2164–2174.
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375.
- Larsson, P.; Engqvist, H.; Biermann, J.; Werner Rönnerman, E.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 2020, 10, 5798.
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322.
- Rothenaigner, I.; Hadian, K. Brief Guide: Experimental Strategies for High-Quality Hit Selection from Small-Molecule Screening Campaigns. SLAS Discov. 2021, 7, 851–854.
- Tanoli, Z.; Aldahdooh, J.; Alam, F.; Wang, Y.; Seemab, U.; Fratelli, M.; Pavlis, P.; Hajduch, M.; Bietrix, F.; Gribbon, P.; et al. Minimal information for chemosensitivity assays (MICHA): A next-generation pipeline to enable the FAIRification of drug screening experiments. Brief Bioinform. 2022, 23, bbab350.
- Rizzuti, B.; Lan, W.; Santofimia-Castaño, P.; Zhou, Z.; Velázquez-Campoy, A.; Abián, O.; Peng, L.; Neira, J.L.; Xia, Y.; Iovanna, J.L. Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules 2021, 11, 1453.
- Choudhary, S.; Lopus, M.; Hosur, R.V. Targeting disorders in unstructured and structured proteins in various diseases. Biophys. Chem. 2022, 281, 106742.
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488.
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: C-Myc as Model System. Cancer Inform. 2017, 16, 1176935117699408.
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018, 3, 5.
- Na, I.; Choi, S.; Son, S.H.; Uversky, V.N.; Kim, C.G. Drug Discovery Targeting the Disorder-To-Order Transition Regions through the Conformational Diversity Mimicking and Statistical Analysis. Int. J. Mol. Sci. 2020, 21, 5248.
- Kim, M.Y.; Na, I.; Kim, J.S.; Son, S.H.; Choi, S.; Lee, S.E.; Kim, J.H.; Jang, K.; Alterovitz, G.; Chen, Y.; et al. Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 2019, 5, eaav9810.
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134.
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442.
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488.
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121.
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848.
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732.
- Mishra, P.; Choudhary, S.; Mukherjee, S.; Sengupta, D.; Sharma, S.; Hosur, R.V. Molten globule nature of Plasmodium falciparum P2 homo-tetramer. Biochem. Biophys. Rep. 2015, 1, 97–107.
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell Mol Life Sci. 2020, 77, 1695–1707.
- Pietrobono, D.; Giacomelli, C.; Trincavelli, M.L.; Daniele, S.; Martini, C. Inhibitors of protein aggregates as novel drugs in neurodegenerative diseases. Glob. Drugs Ther. 2017, 2, 1–5.
- Save, S.S.; Rachineni, K.; Hosur, R.V.; Choudhary, S. Natural compound safranal driven inhibition and dis-aggregation of α-synuclein fibrils. Int. J. Biol. Macromol. 2019, 141, 585–595.
More