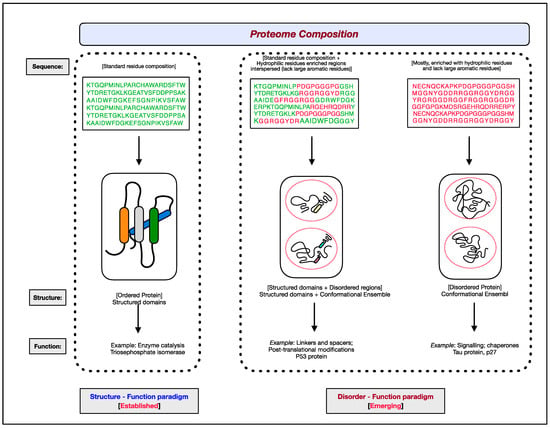
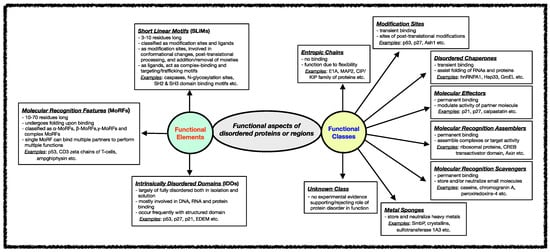
3. Experimental Approaches for Assessing Intrinsic Protein Disorder
Intrinsic protein disorder can be recognized and characterized by various direct and indirect (bio)physical methods. In contrast to direct techniques, which provide structural information about proteins, indirect approaches do not offer any structural details. Still, they suggest a behavior from which the disordered nature of the proteins can be inferred.
3.1. Indirect Methods
Early understanding of the intrinsic structural disorders of proteins was based on a few simple techniques. In general, these indirect intrinsic disorder-identification approaches can quickly provide ample insight into the structural states of a protein or its segments.
Because of the unusual amino acid composition and lack of a compact hydrophobic core, disordered proteins are evident during the purification process. Usually, the molecular mass (M
w) of IDPs estimated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) is higher by a factor of 1.2–1.8 in comparison with that measured by mass spectrometry
[21][29]. Indeed, due to the enrichment of acidic residues and extension in solution, IDPs bind less to SDS and migrate more slowly on the gel in comparison with globular proteins
[91][208]. The aberrant mobility of IDPs is also observed in size-exclusion chromatography (SEC) or gel-filtration (GF) experiments, as a result of which the apparent M
w of proteins with disordered regions is higher
[37][45]. Furthermore, the flexible regions of proteins are known to have increased sensitivity to proteolytic degradation. IDPs, which are more affected than ordered proteins, based on limited in vitro proteolysis, exhibit high inherent flexibility
[21][34][37][64][29,42,45,72].
Other peculiar biochemical behaviors of IDPs/IDRs include insensitivity to high temperatures and stability under acidic treatment. The resistance of IDPs/IDRs to boiling temperatures and acidic pH values has been ascribed to their lower contents of hydrophobic residues and enrichment of polar/charged residues, respectively
[92][93][94][209,210,211]. Neutralizing acidic groups at lower pH levels reduces the net charge on IDPs/IDRs, leading to their increased solubility and a more compact structural state
[37][45]. In contrast to IDPs/IDRs, the aggregation/precipitation of globular/ordered proteins occur at elevated temperatures and under low-pH conditions. While high-temperature conditions expose the hydrophobic core of ordered proteins, acidic conditions cause protonation of their negatively charged side chains, leading to charge imbalances, followed by the disruption of salt bridges and aggregate formation
[21][29].
3.2. Direct Methods
Several techniques provide both steady-state and dynamic structural information on IDPs/IDRs at the residue level. These methods capitalize on the significantly distinct conformational behavior of IDPs compared with that of globular proteins
[21][29]. Some of the most commonly used direct methods are as follows:
3.2.1. X-ray Crystallography
The diffraction intensity and X-ray pattern scattered by electrons in the protein structure are used to construct a three-dimensional (3-D) model of electron density, which, in turn, is used to deduce the atomic nuclei positions in the protein molecule
[21][29]. Disordered regions in X-ray structures appear as missing regions
[20][28]. This method can provide protein structure resolution down to 1Å. Still, additional experimental support is required to be certain about the structural disorder, as missing electron density regions can also result from technical failures in crystallography
[95][212].
3.2.2. Circular Dichroism (CD)
Circular dichroism (CD) is an absorption spectroscopy-based approach that relies on measuring the difference in the absorption spectra of right-handed and left-handed circularly polarized light. Optically active chiral molecules preferentially absorb either right-handed or left-handed circularly polarized light. Near-UV (250–350 nm) and far-UV (190–230 nm) CD signals are generally used to determine different aspects of the structure of proteins in solutions. The near-UV CD spectrum represents the tertiary structure around aromatic residues Phe, Tyr, and Trp
[96][97][213,214]. While intense and detailed spectra characterize ordered proteins, those of IDPs are of low intensity and low complexity. The far-UV CD spectra of the secondary structural elements of proteins are quite distinct; therefore, they are used to determine the proportion of ⍺-helix, β-sheet, turn, PPII helix, and coil conformations in proteins
[98][215]. If the far-UV CD spectrum is indicative of predominantly coil conformations, it indicates the disordered nature of the protein. In the case of proteins having both disordered and ordered regions, the CD does not provide clear information, as it lacks residue-specific details
[20][28].
3.2.3. Nuclear Magnetic Resonance (NMR)
NMR is the most common quantitative technique used for studying IDPs. The spinning ability of the charged atomic nuclei forms the basis of the 3-D structure determination of proteins in solutions using NMR. The directions of these spins are random, but the application of the external magnetic field can align these nuclei in directions either parallel or antiparallel to the applied magnetic field. These two states of nuclei have different energy levels, a low-energy state and a high-energy state. The low-energy state attains a high-energy state upon irradiation with electromagnetic radiation, and free inductive decay (FID) is obtained as the nuclei undergo relaxation. Fourier transformation of FID results in a NMR spectrum with peaks from different types of nuclei in the molecule, which, in turn, is used to characterize the local covalent and spatial arrangement of atoms
[21][39][99][100][29,47,216,217].
In a protein, each nucleus of the individual residues experiences a different magnetic field depending on its microenvironment (referred to as the ‘shielding effect’ or ‘chemical shift’). The chemical shift of the peptide backbone (
1H
⍺,
13CO,
13C
⍺, and
13C
β) can be used to determine the secondary structure type of the given peptide segment
[101][102][218,219]. Amino acids in ordered proteins are packed in different kinds of chemical environments, as a result of which their NMR spectrum resembles a combination of spectra of various secondary structure elements. In contrast, the NMR spectra of disordered proteins with extensive conformational averaging appear as a summation of the random coil spectra of residues of proteins
[99][101][216,218]. In addition to the fine structural details, NMR also provides specific information at the residue level
[20][28].
3.2.4. Small-Angle X-ray Scattering (SAXS)
The SAXS technique can quickly define the structural characteristics of proteins of sizes ranging from a few kilo-Daltons to several giga-Daltons under various experimental setups
[103][104][105][106][220,221,222,223]. Briefly, this method involves exposing samples placed in quartz capillary tubes to a collimated monochromatic X-ray beam source and capturing scattered photons with a detector
[107][224]. Comparative analysis of the electron density distributions of the protein sample and pure solvent/buffer is then conducted to determine various parameters of the proteins in the solution, such as the molecular mass, volume, radius of gyration, folding state, etc.
[103][220]. Moreover, SAXS data can also be used to define protein flexibility and the intrinsically disordered state of proteins in solutions
[106][108][223,225]. The scattering profiles of the proteins obtained from SAXS experiments are most commonly represented as Kratky plots (s
2I(s) as a function of s, where s and I represent the momentum transfer function and scattering intensity), which are used to obtain structural insights into the protein.
In contrast to globular proteins’ bell-shaped Kratky’s plot with well-defined maxima, disordered protein-specific Kratky plots exhibit a plateau for a given range of the momentum transfer function (s), followed by a monotonic increase
[109][110][226,227]. Additionally, the experimentally determined radius of gyration (R
g) of IDPs from the SAXS curve can be directly compared with the theoretical or experimental R
g values of a globular and random coil for a given number of residues. The R
g values of IDPs lie between those of highly compact globular proteins (lowest R
g values) and completely disordered/unfolded proteins represented by random coils (highest R
g values)
[111][228]. Altogether, this method offers fast structural characterization of proteins in solutions with a relatively easy sample preparation protocol and can capture data under near-native conditions
[112][113][229,230]. As the sensitivity of SAXS depends on the particle size, prior removal of the macromolecular aggregates during sample preparation using a method such as sedimentation or size-exclusion chromatography is suggested
[114][231].
Finally, it is worth mentioning that SAXS-based studies of IDPs can use valuable a priori complementary information from several other experimental and in silico protein structure determination methods. For instance, X-ray crystallography depicts the structured regions of a protein, while SAXS defines the protein segments with missing electron density
[115][232]. Similarly, NMR provides information about different domains/complex sub-units during analyses of bimolecular complexes and multi-domain proteins, and SAXS defines their relative inter-domain positions
[116][233]. Furthermore, other complementary techniques, such as CD, spectroscopy, chromatography, etc., and SAXS, can be used for the biophysical characterization of IDPs
[117][234]. A low-resolution protein structure defined through the ab initio modeling of SAXS data alone can be further refined using inputs from protein structure prediction tools, such as I-TASSER, CORAL, etc.
[118][119][235,236]. Recently, various protein structure determination/prediction techniques and SAXS have been used to characterize partially disordered mycobacterial ESX-secretion-associated protein K (EspK)
[120][237].
3.2.5. Cryo-Electron Microscopy (Cryo-EM)
In the last five years, the research area involving the structural characterization of proteins and other biological entities has been revolutionized by the development of cryo-electron-microscopy-based techniques
[121][122][123][124][238,239,240,241]. These methods overcome the limitations of primary methods, i.e., X-ray crystallography and NMR, and allow the structural characterization of relatively large, structurally heterogeneous, flexible, and dynamic assemblies at sub-nanometer atomic resolution (below 4 Å)
[124][125][126][241,242,243]. Typically, a cryo-EM workflow contains three main steps: (a) vitrification (rapid cooling without ice crystal formation) of specimens in an aqueous solution, (b) image acquisition at a low electron dose using electron microscopy, and (c) 3D model reconstruction and validation. Single-particle analysis (SPA) and sub-tomogram averaging (STA) models are most commonly used for the structural annotation of proteins
[127][244]. However, while the globular/ordered/structured regions of proteins can be structurally resolved using cryo-EM, the predicted intrinsically disordered regions in the proximity of flexible regions escape structural assignment
[128][245]. Therefore, similar to x-ray crystallography, a high degree of intrinsic disorder restricts the implementation of cryo-EM techniques. Alternatively, the structure and dynamics of IDPs/IDRs can be investigated by complementing higher-resolution NMR studies of IDRs with the modeling capabilities of cryo-EM
[129][130][246,247]. In conclusion, 3D cryo-EM maps in conjunction with high-resolution data from NMR can model IDPs under physiologically relevant conditions and provide insights into their functional behavior
[126][243].
4. Computational Tools for Disorder Prediction
The biased amino acid compositions and peculiar sequence characteristics of IDPs/IDRs have encouraged the development of various reliable computational tools for studying intrinsic protein disorders. As a result, disorder predictors have been grouped into three distinct classes based on the underlying concepts.
4.1. Propensity-Based Predictors
In principle, a disorder predictor is classified as propensity-based if it depends on some essential physical or chemical characteristics of residues or on prior knowledge of the biological background of intrinsic protein disorder. Disorder-predicting tools, such as FoldIndex, NORSp, GlobPlot, CH plot, and PreLink belong to this category
[37][128][129][130][131][132][133][134][45,245,246,247,248,249,250,251].
4.2. Machine Learning Algorithms (MLAs) Based Predictors
This class of advanced predictors relies on algorithms trained on data sets of experimentally characterized disordered regions and can differentiate disorder and order encoding sequences
[21][29]. Currently, the experimentally characterized disordered proteins are publicly available on three databases: MobiDB (
http://mobidb.bio.unipd.it/; accessed on 7 November 2022), IDEAL (
https://ngdc.cncb.ac.cn/databasecommons/database/id/198; accessed on 7 November 2022), and DisProt (
http://www.disprot.org/; accessed on 7 November 2022)
[84][135][136][92,252,253]. PONDR, Spritz, DisEMBL, RONN, and DISOPRED are a few predictors that fit into this category
[137][138][139][140][141][142][254,255,256,257,258,259].
Recently, the field of protein structure prediction has been revolutionized by the development of the deep learning-based method AlphaFold
[143][260]. This software generates a per-residue confidence score (pLDDT) based on the protein’s amino acid sequence. The most recent version of this tool, i.e., Alphafold2, has been reported to achieve protein structure prediction accuracy competitive with that of experimental determination
[144][145][146][261,262,263]. However, this program gives a low confidence score (pLDDT < 50) for intrinsically unstructured or disordered proteins/regions, and the inconclusive predicted structure resembles a ribbon. In addition, this method does not anticipate the relative likelihood of diverse IDP conformations and the folding pathways followed by IDPs/IDRs attaining an ordered structure upon interaction with other biomolecules
[62][147][70,264]. At present, the AlphaFold Protein Structure Database is considered as the most complete and precise representation of the human proteome
[148][149][265,266].
4.3. Inter-Residue Contact-Based Predictors
Predictors based on the idea that IDPs/IDRs are disordered because they cannot make enough inter-residue contacts required to compensate for the loss of configurational entropy during folding are grouped together as inter-residue contact-based predictors. The above conclusions may be derived by either simple statistics involving contact numbers or through sophisticated techniques of determining the total stabilization energy of a protein. Computational tools, such as IUPred, FoldUnfold, and Ucon belong to this class
[150][151][152][153][267,268,269,270].
At present, there is no “best” disorder prediction computational tool. Therefore, to avoid the limitations of a given tool, prediction results from different disorder predictors relying on distinct principles should be combined to provide a consensus prediction, as implemented by meta-predictors (for example, PONDR-FIT)
[154][271]. Alternatively, publicly available meta-servers (for example, MeDor and metaPRDOS can also be used for quick and simultaneous analysis of protein disorder using multiple predictors
[155][156][272,273].
In several recent articles, extensive comparisons of various computational disorder prediction methods’ performance and comprehensive online resources useful for studying IDPs/IDRs were provided
[157][158][159][274,275,276].
5. Evolution of IDPs/IDRs
The evolution of proteins involves changes in the form of insertions, deletions, or substitutions in their amino acid sequences. Over time, such changes can accumulate in the proteins, giving rise to taxonomic classes having substantial differences in their amino acid compositions
[160][277]. In general, the structure and function of proteins are well conserved, but several exceptions exist. Several previous studies suggested that, even if the protein sequence diverges extensively, the protein function is well-conserved
[161][278]. Hence, proteins are generally considered as the ‘chemical fingerprints’ of evolutionary history, as they manifest the underlying genetic changes as amino acid sequences.
The evolution of intrinsic disorder exhibits a wavy pattern in which highly disordered primordial proteins with predominantly RNA-chaperone-like activities were slowly replaced with highly structured proteins
[162][163][118,279]. Later, because of its peculiar features regarding the regulation of complex cellular processes, protein disorder was reinvented at various succeeding evolutionary stages, resulting in the creation of more complex organisms from the last universal ancestor
[164][165][280,281].
Several mechanisms, such as de novo generation, horizontal gene transfer, and lateral gene transfer, can give rise to genes that encode IDPs
[49][166][57,282]. Approximately 14% of Pfam domains, predicted to be mostly disordered and shared by many protein families, appear to have originated from domain duplications and module exchange between genes
[167][196]. The high frequency of occurrence of tandemly repeated sequences in IDPs/IDRs suggests that the expansion of internal repeat regions (microsatellite and minisatellite coding regions) is another possible way by which the IDPs encoding genes arose
[168][169][283,284]. Looking at the exceptional functional variability conferred to IDPs/IDRs due to the genetic instability of repetitive elements, the mechanism of the extension of repeat elements appears as the frequent method of disorder spread during evolution and rapid genomic changes in adaptation
[28][170][171][36,285,286]. Furthermore, these IDPs/IDRs can also act as hot spots for mutations, leading to the loss of different functional modalities and thus resulting in various types of diseases, including cancer
[172][173][174][287,288,289]. Seera and Nagarajaram have recently shown that the disease-causing missense mutations within IDRs reduce the overall conformation heterogeneity of the IDRs as compared to their wild type counterparts, and the few ‘locked’ dominant conformations presumably limit their interaction with the cognate partners
[175][290].
Recent studies have shown that disordered protein segments are encoded by GC-enriched gene regions, which, in turn, directly correspond to the disorderedness of the encoded proteins
[176][177][291,292]. This GC enrichment is due to the prevalence of amino acids coded by GC-rich codons (G, A, R, and P) in the disordered regions of proteins
[176][291]. At the residue level, a relatively higher rate of evolutionary changes in the disordered regions of proteins was observed compared with that in the ordered/globular domains, as there were no structural constraints to maintaining a 3-D structure
[178][293]. However, in certain cases, structured domains and disordered regions of proteins have been observed to co-evolve at higher rates
[179][180][294,295]. Despite these rapid changes, the biological functions of the structured domains and disordered regions are always conserved
[181][296]. Hence, a deeper understanding of the conformation ensemble–function relationship will help to decipher the evolutionary trajectory of IDPs.
Based on the conservation of sequences coding for protein disorder, disordered residues have been classified as constrained (both sequence and disorder are conserved) or flexible (only protein disorder is conserved). Together, constrained and flexible disorder residues are known as conserved disorder. On the other hand, if neither disorder nor the residues encoding it are conserved, such a disorder class is known as non-conserved disorder
[182][297]. This integrated structural and evolutionary approach has recently been used to define the determinants of the functional adaptability of the neutrophin family of proteins involved in neuronal development
[183][298].
Considering that the disordered regions in proteins have a distinct amino acid composition and evolutionary rate as compared with that of ordered regions, the substitution frequencies of residues in the disordered regions must also be distinct from those found in ordered regions. Thus, identifying the evolutionary and functional features of IDPs/IDRs has become a computational challenge, as most of the sequence analysis tools and parameter optimization procedures are aimed at ordered/structured regions of proteins. Recently, methods evaluating disordered proteins’ molecular features and sequence composition in a position-specific manner have been developed. These advancements have allowed researchers to pursue alignment-based evolutionary studies on IDPs/IDRs without aligning the residues discretely
[184][185][186][299,300,301].
6. IDPs/IDRs in Diseases
Like structured proteins, the expression, localization, and interactions of intrinsically disordered proteins (IDPs) are also highly coordinated and regulated. Multiple checkpoints at various stages of the expression of IDPs-specific genes (from transcript synthesis to protein degradation) ensure the availability of IDPs in appropriate quantities and for the desired duration, preventing any ectopic interactions
[187][302]. Several studies have shown the role of IDPs/IDRs in different human disorders, including diabetes, cancer, amyloidosis, neurodegenerative, and cardiovascular diseases
[188][189][303,304]. Some well-studied examples of IDPs associated with human disease are p53, Mdm2, PTEN, c-Myc, AF4, BRCA1, EWS, Bcl-2, c-Fos, HPV oncoproteins, etc.
[188][190][191][192][303,305,306,307]. Moreover, the deposition of ⍺-synuclein, tau, and amyloid-β proteins leads to Alzheimer’s disease, the accumulation of ⍺-synuclein results in Parkinson’s disease, and aggregates of PrP
SC cause prion diseases. The expansion of CAG triplet repeats in disease genes, which introduces disorder, results in the family of polyQ diseases, such as Kennedy’s disease, Huntington’s disease, etc.
[193][194][195][196][197][198][199][308,309,310,311,312,313,314].
In the last two decades, the role of IDPs in human diseases has been actively studied, giving rise to new mechanistic findings that have led to the formation of the D
2 concept (‘Disorder to Disorders’)
[188][303]. Several comprehensive reviews and thematic series articles have been published covering the significance of IDPs in diseases
[200][201][202][315,316,317]. For instance, Coskuner and Uversky described various hypotheses proposed to explain the molecular mechanisms of the pathogenesis of Alzheimer’s and Parkinson’s diseases and suggested the need for the development of new techniques through the integration of quantum and statistical mechanics, thermodynamics, bioinformatics, and machine learning approaches, which, in turn, may lead to the development of new experimental approaches
[203][204][205][206][207][208][209][210][318,319,320,321,322,323,324,325]. However, at present, there are several limitations and challenges associated with in silico studies of IDP-associated neurodegenerative disorders
[211][212][326,327]. Another study found that an NADH-stabilized 26S proteasomal complex could degrade IDPs efficiently. Therefore, the accumulation of disease-causing disordered proteins, such as tau, c-Fos, p53, etc., can be prevented by the selective degradation of IDPs in an ATP-independent manner
[213][328]. Moreover, the analysis of components of the ATP-dependent ubiquitin-proteasome degradation system (UPS) revealed the importance of the disorder content and MoRFs of the complex in neurodegenerative disorders and cancers
[214][329]. However, identifying key mutations, PTM sites, and functional motifs in the disordered regions, exploring the evolutionary history of IDPs involved in diseases, understanding the cooperative functioning of ordered and disordered domains, and dissecting the IDPs’ interactome are some of the many active research areas involving IDPs/IDRs and diseases
[173][215][216][217][218][219][288,330,331,332,333,334].
7. IDPs/IDRs as Drug Targets
With increasing evidence of their involvement in molecular functions complementing globular domains, essential biological processes, protein–nucleic acid interactions, protein–protein interactions, and diseases, IDRs/IDPs have emerged as one of the prime targets for drug discovery or repurposing
[220][221][222][223][224][225][142,335,336,337,338,339]. However, IDP characteristics, such as a lack of a sTable 3D structure, very high flexibility, conformational ensembles, susceptibility to proteolytic cleavage, protein aggregation, etc., limit the application of the most-established experimental assays and computational methods that would otherwise work for ordered/globular proteins
[226][227][228][229][230][340,341,342,343,344]. Therefore, IDP-specific drug screening/development is mainly a tradeoff between binding affinity/specificity and the alternation in the functioning of disordered proteins with other features, such as solubility, crowding, efflux, metabolism, etc., a potentially relevant role
[231][345].
Broadly, disordered proteins/regions have been used in drug development procedures by targeting their conformational changes, interactions, and self-aggregating behavior
[232][346]. For example, the inhibitor 10058F4 of Myc proto-oncogene protein (MYC) binds to MYC and prevents conformational disorder-to-order transition, which, in turn, blocks MYC-MAX complex-driven tumorigenesis
[25][233][234][235][236][33,347,348,349,350]. Similarly, Methyl-CpG-binding domain protein 2 (MBD2) inhibitors restrict the folding of MBD2 upon binding to its partner p66α. This MBD2-p66α is known to regulate the Mi-2/NuRD chromatin remodeling complex involved in promoting metastasis in various cancer cells through epithelial–mesenchymal transition (EMT)
[237][238][351,352]. In contrast to ordered proteins, the protein–protein interactions involving IDPs offer uneven, shorter, compact, and more mimicable surfaces for the tighter binding of small drug molecules
[239][240][241][353,354,355]. In recent times, potential drug molecules have been designed to target either the disordered segment or the binding region of the interacting molecule. For instance, nutlins binding to Mdm2 prevent the interaction of Mdm2 with the disordered regions of p53, which activates the p53 pathway, leading to apoptosis, cell-cycle arrest, and the inhibition of the uncontrolled cell growth of human tumor xenografts
[242][356]. Additionally, an FDA-approved compound, trifluoperazine dihydrochloride, was found to bind to a disordered region of multifunctional protein nuclear protein 1 (NUPR1) and arrest pancreatic ductal adenocarcinoma (PDAC) development
[243][357]. Moreover, the disordered proteins from pathogens can also be targeted to interrupt their interaction with host proteins, which they utilize for their survival and pathogenesis
[244][358]. In a recent review, Santofimia et al. comprehensively described targeting IDPs in various protein–protein and protein–nucleic acid interactions involved in cancer
[245][359]. Furthermore, compounds, such as curcumin, rosmarinic acid, ferulic acid, and safranal, have also been reported to prevent the aggregation of α-synuclein protein by binding to monomers, thus inhibiting the polymerization of these proteins, which results in various neuronal malignancies
[246][247][360,361]. In summary, deciphering the sequence–ensemble–function relationship of IDPs/IDRs and the development of efficient computational modeling approaches will help to unravel the enormous potential of disordered proteins as drug targets.