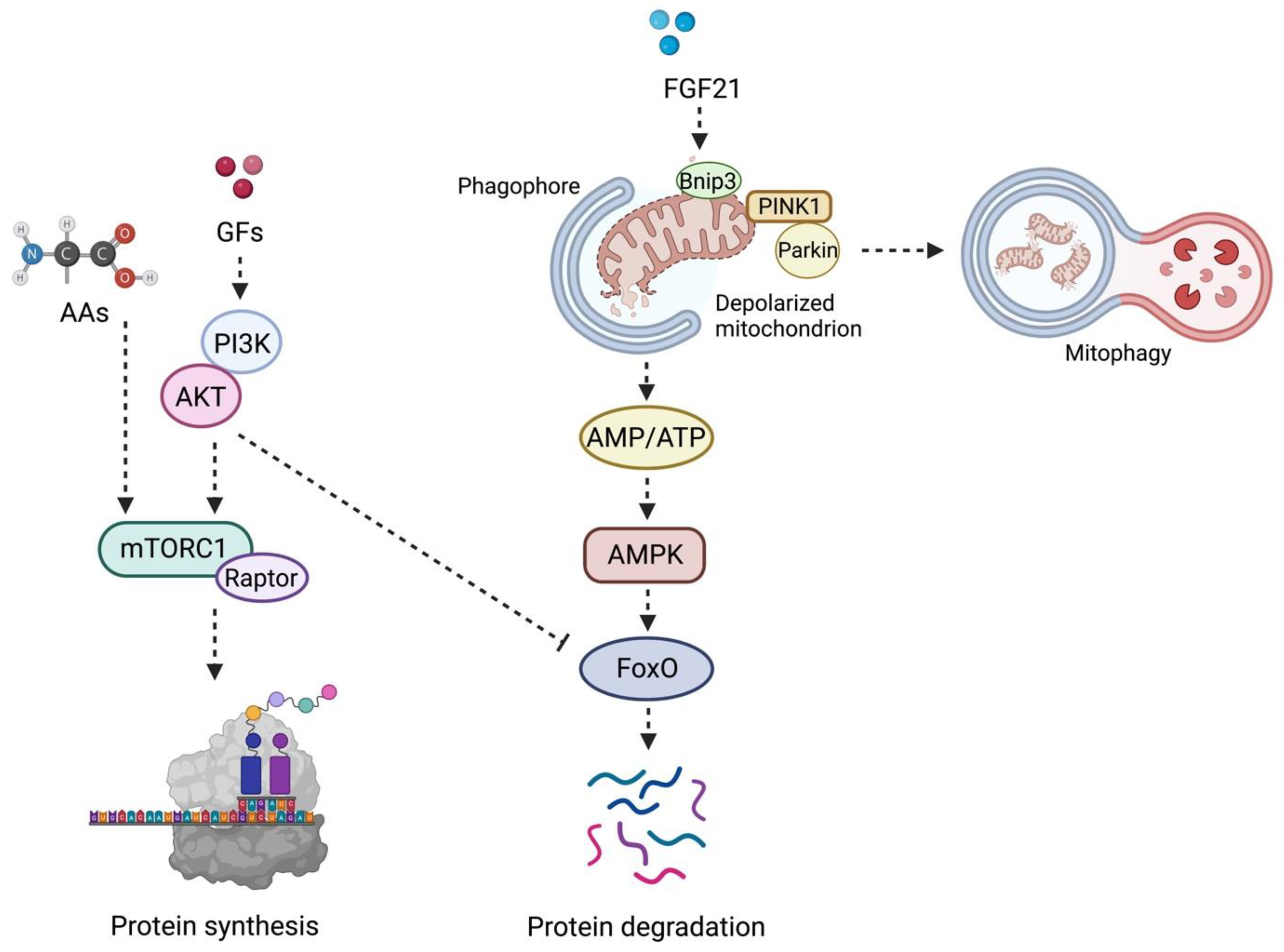
Sarcopenia, the age-related decline of muscle mass and strength/function is a major risk factor for disability and loss of independence in late life. Studies have shown that behavioral interventions (e.g., physical activity, adapted nutrition) reduce the rate of muscle wasting during aging. However, an incomplete understanding of the mechanisms driving age-related muscle loss has hampered the development of effective drugs to prevent or treat sarcopenia. Altered muscle protein metabolism is considered to be one of the main factors underlying the development and progression of sarcopenia. While basal rates of muscle protein synthesis (MPS) and degradation (MPD) seem to be unaffected by age, the anabolic response to a variety of stimuli (e.g., exercise, nutrient ingestion) is blunted during aging. The mammalian target of rapamycin (mTOR) is a key regulator of muscle anabolic and catabolic pathways and, hence, a promising target for interventions against sarcopenia.
- aging
- skeletal muscle
- autophagy
- mitophagy
- protein synthesis
- protein degradation
1. Supramolecular Organization of the Mammalian Target of Rapamycin
2. Muscle Protein Synthesis: The Mammalian Target of Rapamycin Complex 1 Axis
mTORC1 Axis: Different Stimuli for Muscle Protein Synthesis
3. Muscle Protein Degradation: The Other Face of the mTORC1 Axis during Aging
References
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Aylett, C.H.S.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of Human mTOR Complex 1. Science 2016, 351, 48–52.
- Yu, Z.; Chen, J.; Takagi, E.; Wang, F.; Saha, B.; Liu, X.; Joubert, L.M.; Gleason, C.E.; Jin, M.; Li, C.; et al. Interactions between mTORC2 Core Subunits Rictor and MSin1 Dictate Selective and Context-Dependent Phosphorylation of Substrate Kinases SGK1 and Akt. J. Biol. Chem. 2022, 298, 102288.
- Scaiola, A.; Mangia, F.; Imseng, S.; Boehringer, D.; Berneiser, K.; Shimobayashi, M.; Stuttfeld, E.; Hall, M.N.; Ban, N.; Maier, T. The 3.2-Å Resolution Structure of Human mTORC2. Sci. Adv. 2020, 6, eabc1251.
- Baraldo, M.; Nogara, L.; Dumitras, G.A.; Dondjang, A.H.T.; Geremia, A.; Scalabrin, M.; Türk, C.; Telkamp, F.; Zentilin, L.; Giacca, M.; et al. Raptor Is Critical for Increasing the Mitochondrial Proteome and Skeletal Muscle Force during Hypertrophy. FASEB J. 2021, 35, e22031.
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of Muscle Atrophy and Hypertrophy: Implications in Health and Disease. Nat. Commun. 2021, 12, 330.
- Baehr, L.M.; Hughes, D.C.; Waddell, D.S.; Bodine, S.C. SnapShot: Skeletal Muscle Atrophy. Cell 2022, 185, 1618–1618.e1.
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 5759.
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The Neuromuscular Junction Is a Focal Point of MTORC1 Signaling in Sarcopenia. Nat. Commun. 2020, 11, 4510.
- Baraldo, M.; Geremia, A.; Pirazzini, M.; Nogara, L.; Solagna, F.; Türk, C.; Nolte, H.; Romanello, V.; Megighian, A.; Boncompagni, S.; et al. Skeletal Muscle mTORC1 Regulates Neuromuscular Junction Stability. J. Cachexia Sarcopenia Muscle 2020, 11, 208–225.
- Ketilly, P.; Alves, N.; Cruz, A.; Silva, W.J.; Labeit, S.; Moriscot, A.S. MiR-29c Increases Protein Synthesis in Skeletal Muscle Independently of AKT/mTOR. Int. J. Mol. Sci. 2022, 23, 7198.
- Jin, J.; Li, F.; Fan, C.; Wu, Y.; He, C. Elevated Mir-145-5p Is Associated with Skeletal Muscle Dysfunction and Triggers Apoptotic Cell Death in C2C12 Myotubes. J. Muscle Res. Cell Motil. 2022, 43, 135–145.
- Picca, A.; Calvani, R.; Sirago, G.; Coelho-Junior, H.J.; Marzetti, E. Molecular Routes to Sarcopenia and Biomarker Development: Per Aspera Ad Astra. Curr. Opin. Pharmacol. 2021, 57, 140–147.
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203.
- Nagao, H.; Cai, W.; Albrechtsen, N.J.W.; Steger, M.; Batista, T.M.; Pan, H.; Dreyfuss, J.M.; Mann, M.; Kahn, C.R. Distinct Signaling by Insulin and IGF-1 Receptors and Their Extra- And Intracellular Domains. Proc. Natl. Acad. Sci. USA 2021, 118, e2019474118.
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101.
- Philp, A.; Hamilton, D.L.; Baar, K. Highlighted Topic Signals Mediating Skeletal Muscle Remodeling by Activity Signals Mediating Skeletal Muscle Remodeling by Resistance Exercise: PI3-Kinase Independent Activation of mTORC1. J. Appl. Physiol. 2011, 110, 561–568.
- Frey, J.W.; Jacobs, B.L.; Goodman, C.A.; Hornberger, T.A. A Role for Raptor Phosphorylation in the Mechanical Activation of mTOR Signaling. Cell. Signal. 2014, 26, 313–322.
- You, J.S.; Mcnally, R.M.; Jacobs, B.L.; Privett, R.E.; Gundermann, D.M.; Lin, K.H.; Steinert, N.D.; Goodman, C.A.; Hornberger, T.A. The Role of Raptor in the Mechanical Load-Induced Regulation of mTOR Signaling, Protein Synthesis, and Skeletal Muscle Hypertrophy. FASEB J. 2019, 33, 4021–4034.
- Vary, T.C.; Jefferson, L.S.; Kimball, S.R. Amino Acid-Induced Stimulation of Translation Initiation in Rat Skeletal Muscle. Am. J. Physiol. 1999, 277, 1077–1086.
- Jeyapalan, A.S.; Orellana, R.A.; Suryawan, A.; O’Connor, P.M.J.; Nguyen, H.V.; Escobar, J.; Frank, J.W.; Davis, T.A. Glucose Stimulates Protein Synthesis in Skeletal Muscle of Neonatal Pigs through an AMPK- and mTOR-Independent Process. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 595–603.
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science 2008, 320, 1496–1501.
- Korzick, D.H.; Sharda, D.R.; Pruznak, A.M.; Lang, C.H. Aging Accentuates Alcohol-Induced Decrease in Protein Synthesis in Gastrocnemius. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R887–R898.
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 Underlies Age-Related Muscle Fiber Damage and Loss by Inducing Oxidative Stress and Catabolism. Aging Cell 2019, 18, e12943.
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 Causes Coupled Loss of Desmin, Z-Bands, and Thin Filaments in Muscle Atrophy. J. Cell Biol. 2012, 198, 575–589.
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 Ubiquitin Ligase TRIM32 Regulates Myoblast Proliferation by Controlling Turnover of NDRG2. Hum. Mol. Genet. 2015, 24, 2873–2883.
- Skoglund, E.; Grönholdt-Klein, M.; Rullman, E.; Thornell, L.E.; Strömberg, A.; Hedman, A.; Cederholm, T.; Ulfhake, B.; Gustafsson, T. Longitudinal Muscle and Myocellular Changes in Community-Dwelling Men over Two Decades of Successful Aging–The ULSAM Cohort Revisited. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 654–663.
- Kenyon, C.J. The Genetics of Ageing. Nature 2010, 464, 504–512.
- Sirago, G.; Picca, A.; Giacomello, E.; Marzetti, E.; Toniolo, L. The Contribution of Genetics to Muscle Disuse, Retraining, and Aging. Genes 2022, 13, 1378.
- Segalés, J.; Perdiguero, E.; Serrano, A.L.; Sousa-Victor, P.; Ortet, L.; Jardí, M.; Budanov, A.V.; Garcia-Prat, L.; Sandri, M.; Thomson, D.M.; et al. Sestrin Prevents Atrophy of Disused and Aging Muscles by Integrating Anabolic and Catabolic Signals. Nat. Commun. 2020, 11, 189.
- Gu, X.; Jouandin, P.; Lalgudi, P.V.; Binari, R.; Valenstein, M.L.; Reid, M.A.; Allen, A.E.; Kamitaki, N.; Locasale, J.W.; Perrimon, N.; et al. Sestrin Mediates Detection of and Adaptation to Low-Leucine Diets in Drosophila. Nature 2022, 608, 209–216.
- Xu, D.; Shimkus, K.L.; Lacko, H.A.; Kutzler, L.; Jefferson, L.S.; Kimball, S.R. Evidence for a Role for Sestrin1 in Mediating Leucine-Induced Activation of mTORC1 in Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E817–E828.