Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ornella Franzese and Version 2 by Rita Xu.
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) induce cytotoxic effects as single agents in tumors characterized by defective repair of DNA double-strand breaks deriving from BRCA1/2 mutations or other abnormalities in genes associated with homologous recombination. Preclinical studies have shown that PARPi-induced DNA damage may affect the tumor immune microenvironment and immune-mediated anti-tumor response through several mechanisms. In particular, increased DNA damage has been shown to induce the activation of type I interferon pathway and up-regulation of PD-L1 expression in cancer cells, which can both enhance sensitivity to Immune Checkpoint Inhibitors (ICIs).
- PARP inhibitor
- BRCA
- DNA damage response
- immunotherapy
1. Introduction
The PARP superfamily comprises 17 different molecules, characterized by distinctive activities [1][2][1,2]. PARP-1, the most representative member of the group, was originally recognized for its function of identifying and repairing DNA single-strand breaks (SSBs) [3]. PARP-1 exerts its activity through the post-translational modification of partner molecules by adding linear or branched poly(ADP-ribose) (PAR) chains using NAD+ as its substrate [4], thus modulating the functional engagement of enzymes involved in the repair of SSBs (i.e., base excision repair (BER)) and the activity of other repair systems [5][6][7][8][9][10][5,6,7,8,9,10]. Moreover, PARP-1 is involved in the regulation of other cellular processes, including gene expression, mitosis, apoptosis and sex hormone-mediated signaling [11][12][11,12]. Besides PARP-1, PARP-2 and PARP-3 are also involved in the repair of DNA damage, presenting a certain degree of redundancy in some of PARP-1 functions [13].
Inhibition of PARP activity has been shown to induce synthetic lethality in breast cancer gene 1 and 2 (BRCA1/2) mutated tumors, which are not able to repair DNA double strand breaks (DSBs) [8][14][15][16][17][18][8,14,15,16,17,18]. Remarkably, BRCA1 and BRCA2 preserve genomic stability and inhibit tumorigenesis by fostering DSBs repair via the homologous recombination (HR) system and are often mutated in hereditary breast and ovarian cancers [19]. Moreover, BRCA1/2 mutations increase the risk for pancreatic and prostate tumors [20][21][22][20,21,22]. PARP inhibitors (PARPi) compete with NAD+ for the binding to the catalytic active PARP domain, inhibiting the synthesis of PAR chains with consequent impairment of chromatin structure remodeling and recruitment of DNA repair proteins at the damaged site. Thus, inhibition of PARP activity results in the generation of persistent SSBs that are converted into DSBs due to collapse of stalled replication forks during DNA replication [23]. In HR-defective cells, replication-associated DSBs cannot be effectively repaired and become lethal [18][24][18,24]. Furthermore, in the absence of an efficient HR-mediated repair of DSBs, aberrant activation of the error-prone nonhomologous end joining (NHEJ) takes place. NHEJ is an alternative DSBs repair system that directly re-ligates DNA broken ends without taking into account sequence homology. This leads to an inadequate repair of DSBs, contributing to genomic instability and tumor cell lethality [25]. In addition, PARPi can “trap” PARP-1/PARP-2 at DNA breaks due to inhibition of PARP auto-PARylation, thus preventing PARP protein release from the site of damage. Moreover, PARPi interaction with the NAD+ site allosterically increases the DNA binding of PARP N-terminal region. This leads to stalling of the replication fork and eventually cell killing mainly as a consequence of SSBs conversion into unrepaired DSBs [26][27][28][26,27,28]. The cytotoxicity based on this mechanism requires PARP-1 expression, since its silencing abolishes both PARPi trapping efficiency and cell killing effects, whereas PARP-2 (whose expression is much lower than PARP-1) has minimal influence on tumor sensitivity to PARPi [26].
The anti-tumor efficacy of PARPi as monotherapy, in the context of tumors expressing either germline or somatic mutations in the BRCA genes, has been established by pre-clinical and clinical investigations that have led to the FDA/EMA approval of four different drugs (i.e., olaparib, rucaparib, niraparib and talazoparib) for the treatment of advanced/metastatic ovarian cancer, triple negative breast cancer (TNBC), pancreatic cancer and prostate cancer [29][30][31][32][33][29,30,31,32,33]. Approved PARPi possess similar ability to inhibit the catalytic activity of PARP-1 and some of them also inhibit PARP-2 and PARP-3. Moreover, they markedly differ in PARP trapping activity with talazoparib being the most potent in this respect [34]. Consistently, talazoparib is the PARPi with the highest ability to induce cytotoxic effects in HR-deficient cells [35].
From the time PARPi have been approved [36][37][38][36,37,38], other genomic defects that result in altered HR function have been found to increase tumor susceptibility to these agents, leading to the extension of their use, in the case of ovarian cancer, regardless of the BRCA mutational status [39][40][41][39,40,41]. However, even in the presence of BRCA mutations or HR deficiency not all patients show a favorable response to PARPi with more than 40% of them experiencing treatment failure. For instance, the phase 3 SOLO-1 trial indicated that the 5-year progression-free survival (PFS) of patients with advanced, newly diagnosed, BRCA mutated ovarian cancer, treated with olaparib as maintenance monotherapy for up to 2 years, was about 50% (48% (95% CI 41–55) vs. 21% (14–28) in the placebo group) [42]. These data highlight not only the need of additional biomarkers to better select patients who might benefit from PARPi as monotherapy, but also the importance of identifying a solid biological rationale for their combination with agents endowed with a non-overlapping mechanism of action.
Immune checkpoint (IC) inhibitors (ICIs), working by antagonizing the immunosuppressive strategies engaged by tumor cells, have strongly impacted the outcome of cancer immunotherapy. Indeed, ICIs have renewed the therapeutic approach of several advanced/metastatic forms of cancer leading to the FDA/EMA authorization of anti-Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) (i.e., ipilimumab), Programmed Cell Death 1 (PD-1) (i.e., nivolumab, pembrolizumab, cemiplimab, dostarlimab), and Programmed Cell Death Ligand 1 (PD-L1) (i.e., atezolizumab, avelumab, durvalumab) monoclonal antibodies (mAbs). Approved indications include advanced/metastatic melanoma, non-small cell lung cancer (NSCLC) or small-cell lung cancer (SCLC), mesothelioma, esophageal squamous cell carcinoma, gastric cancer, hepatocellular carcinoma, urothelial carcinoma, cervical cancer, microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer, renal cell carcinoma, Hodgkin’s lymphoma, head and neck squamous cell carcinoma, Merkel-cell carcinoma, and tumor mutational burden-high cancers [43]. However, while showing significant efficacy, the clinical outcome of IC blockade may differ, with several patients not responding at any rate or finally showing resistance and tumor recurrence [44]. This strongly advocates the requirement of effective combined approaches to overcome resistance.
Remarkably, the efficacy of IC blockade has been shown to be largely conditioned by neoantigen expression and recognition [45][46][47][48][45,46,47,48], suggesting that the permanence of DNA alterations deriving from PARP inhibition could support ICI-mediated boosting of the antitumor response. Interestingly, tumors sensitive to PARPi as a result of deleterious germline or somatic mutations in the BRCA genes have been frequently described as poorly responding to IC blockade single treatment [49][50][49,50]. Moreover, PARPi have demonstrated to control the tumor immune microenvironment (TME) by modulating both cancer cell genomic instability and T cell mediated responses. These observations have provided the rationale for testing the combined inhibition of PARP and ICs for the management of selected tumors.
2. Influence of PARP Inhibition on Immune Response to Tumors
2.1. Well Established Effects of PARP Inhibition on the Different Features of TME
The influence of PARPi on anti-tumor T cell immune response and on TME features can be mediated by different mechanisms. Some of them are directly related to the defective DNA damage repair, others are instead associated with the activation of still not fully clarified pathways, which ultimately convert an immunologically silenced “cold” TME into an immunologically “hot” milieu, characterized by effective CD8+ T cells, type I macrophages (M1) and natural killer (NK) cells (see below). As indicated above, inhibition of PARP activity induces cell death preferentially in BRCA1/2 mutated or otherwise HR-defective tumors which are unable to repair DNA DSBs [30][51][30,51]. In fact, besides BRCA1/2 mutations, a variety of additional genetic alterations may contribute to induce in cancer cells a “BRCAness phenotype”, characterized either by a defective DNA Damage Response (DDR) [52] or by reduced expression of other factors involved in DNA repair (e.g., PI3-kinase-related protein kinases Ataxia-Telangiectasia Mutated (ATM), RAD3-related (ATR), Checkpoint Kinase 1 and 2 (CHK1/2), DSS1, RAD51, CDK12, TP53, or Phosphatase and Tensin Homolog (PTEN)) [52][53][54][55][52,53,54,55]. Therefore, these molecular features may represent responsive predictive biomarkers for PARPi-based treatment of non-BRCA mutated tumors [55][56][57][55,56,57]. On the other hand, among the first evidence about cancer responsiveness to ICIs, a phase 2 investigation showed that the efficacy of the anti-PD-1 pembrolizumab was superior in solid tumors with defective MMR enzymatic activity, and that the clinical outcome was related to the extent of somatic mutations, with best patient’s response occurring in the presence of high frequency of neoantigen-specific T cells [58]. DDR defects, including those affecting BRCA1/2, are related to greater tumor mutational burden in tumors, including ovarian cancer and NSCLC [59][60][59,60], contributing to increase the amount of tumor-specific neoantigens, both valuable biomarkers for predicting the efficacy of ICI therapy, in combination with other indicators [61][62][63][64][65][66][67][68][61,62,63,64,65,66,67,68]. Therefore, the relationship existing amid tumor DNA damage and an improved immune response has suggested a potential role for PARPi as sensitizers of tumor cells to ICIs [69][70][69,70], by impairing DNA repair and producing genomic instability with consequent expression of neoantigens that could support the re-invigoration of anti-tumor T cell mediated response. Neoantigen presentation provided by antigen-presenting cells (APCs), including dendritic cells (DCs), monocytes/macrophages and B lymphocytes, in the context of major histocompatibility complex class I (MHC I) molecules, is required to stimulate the cytotoxic response of CD8+ T cells. Of note, MHC I expression and antigen presentation are improved by DDR [60]. PARP inhibition itself has been shown to stimulate both MHC I expression, following activation of ATM/ATR kinases [71][72][71,72], and DDR, alongside immunogenic cell death (ICD), thus stimulating the damage-associated molecular patterns (DAMPs) to further support the engagement of APCs [73]. Inhibition of PARP-1 increases the extent of DNA damage and the release/accumulation into the cytoplasm of DNA fragments. These can be sensed by innate anti-viral DNA mechanisms, including the cyclic GMP–AMP synthase (cGAS)/stimulator of interferon (IFN) genes (STING) which releases the STING carboxyl terminus to subsequently recruit and activate by phosphorylation the TANK-binding kinase 1 (TBK1) and the IFN regulatory factor 3 (IRF3) [74][75][76][74,75,76]. The cGAS/STING/TBK1/IRF3 pathway is responsible for the activation of type I IFN response [69][77][78][79][69,77,78,79]. In addition, activation of IRF3 is required to produce inflammatory cytokines including IFN-γ, TNF-α, IL-6 [80][81][82][80,81,82], and IL-12, of which the latter is critical in promoting the T cell-DC crosstalk [83] required to promote antigen presentation. Other released proteins include C-X-C motif chemokine ligand (CXCL)1, CXCL2, CXCL9, and CXCL10 that support both development and tumor infiltration of macrophages and CD8+ T cells [80]. STING also stimulates the activation of NF-κB, which collaborates with IRF3 in promoting the release of pro-inflammatory cytokines such as IL-1β, IL-6 and IFN-γ [84][85][86][84,85,86] Furthermore, although type I IFNs are mainly produced by DCs in the TME, a paracrine cGAS/STING pathway stimulation has been observed in neighboring DCs, exclusively in BRCA1-deficient models of TNBC and ovarian cancer, mediated by pro-inflammatory cytokines release and possibly tumor DNA exocytosis [87][88][87,88], according to the described role played by extracellular DNA in the generation of an inflammatory phenotype [89]. Thus, based on the different trapping efficiency and ability to induce DNA damage and generate DNA fragments of the various clinically relevant PARPi, it can be hypothesized that PARPi with strong trapping activity (e.g., talazoparib) will more efficiently synergize with ICIs than weak PARP trappers (e.g., veliparib). Of note, improved antigen presentation and increased frequency of tumor associated DCs with high expression of CD40, CD80, and CD86 co-stimulatory molecules have been described in response to olaparib, together with enhanced transition of CD8+ T cells to the tumor site [90]. The CD40/CD40L engagement has been shown to positively influence both humoral and cellular immune responses [91]. Moreover, the increased CD80/CD86 expression induced by olaparib holds additional implications. In fact, besides representing a critical player in T cell co-stimulation, CD80 forms heterodimers in cis with PD-L1, reducing its own interaction with CTLA-4, while maintaining the ability to activate CD28 [92]. Despite possessing a very high mutational burden, SCLC is frequently characterized by severe immunosuppression and poor intra-tumor T-cell infiltration [93]. According to these observations, clinical studies exploring the use of anti PD-1 or PD-L1 mAbs in SCLC patients have revealed poor clinical outcome [94]. SCLC over-expresses PARP-1 [95], whose inhibition was found to up-regulate intra-tumor T cell infiltration and to synergize with PD-L1 blockade, provoking substantial tumor decrease in mouse models [96]. Conversely, olaparib and anti–PD-L1 single treatments did not induce marked antitumor effects. Remarkably, knockdown experiments have unequivocally established the direct role of the STING/TBK1/IRF3 pathway in the antitumor immune response stimulated by the combined treatment in SCLC [96]. Increasing evidence suggests that the distribution, density, and phenotype of tumor infiltrating lymphocytes (TILs) positively affect the efficacy of ICIs [97]. Different studies have demonstrated that PARP inhibition up-regulates intra-tumor infiltration by both CD4+ and CD8+ T cell subsets [87][98][87,98]. In particular, Strickland et al. established that BRCA1/2-defective high-grade serous ovarian cancers exhibit significantly higher whole CD3+ as well as CD8+ tumor T cell infiltration and better patients’ survival outcome compared with HR-proficient tumors [59]. Of note, olaparib-induced increase in terms of CD8+ T cell recruiting in the TME has been shown to be mediated by the tumor cGAS/STING pathway, an effect more obvious in HR-defective than in HR-proficient TNBC cells [87]. The preferential stimulation of STING/TBK1/IRF3 pathway in BRCA1-defective tumors has been confirmed by further in vivo studies [99], speaking in favor of a prediction for greater intra-tumor T cell penetration in BRCA1-defective cancers and of a more modest effect of combined therapy in tumors with a proficient HR system [86]. Conversely, according to other studies [100], PARP inhibition has been shown to induce cytosolic DNA accumulation and type I IFN response, along with increased intra-tumor CD8+T cell infiltration dependent on C-C chemokine ligand 5 (CCL5) and CXCL10 chemokines, irrespective of the BRCA mutational state. These observations foster the crucial issues of whether: (i) a defective BRCA is strictly required for the STING-dependent immunological improvement associated with PARPi, and (ii) additional mechanisms are involved beside those induced by unsolved genomic defects. However, in BRCA competent cancers, characterized by a possibly reduced STING-associated IFN type I pathway, where DNA lesions induced by PARP blockade could be inadequate, the use of a STING agonist combined with IC blockade has been suggested as an alternative potential therapeutic combination approach [87][101][87,101]. It is also well acknowledged that modulation of the intra-tumor T cell equilibrium toward a CD4+Th1 response is critical to promote cytotoxic CD8+ T cell functional activity and an effective anti-tumor response [102]. CD4+ Th1 effector cells, by producing key cytokines such as IFN-γ, TNF-α, and IL-2, are critical players in anti-tumor protection, able to mediate cancer cell elimination through activation of both innate and adaptive immunity [103]. In the TME, IFN-γ improves the immunogenicity of tumor cells by increasing the expression of MHC class I and II, which makes them more susceptible to recognition by effector cells [104]. Moreover, IFN-γ enhances the tumoricidal action of M1 macrophages and the recruitment of NK and T cells from the periphery to the tumor site via CXCL9 and CXCL10 chemokines. Remarkably, impairment of PARP-1 activity has been shown to bias T cell phenotype balance by favoring Th1 subset differentiation while lowering the Th2 specific immune response [105]. This effect has been possibly related to calpain-mediated degradation of the transcription factor STAT6, which is required for Th2 differentiation signaling [106]. An additional mechanism proposed for PARPi-associated inhibition of Th2 responses is provided by the reduced expression of the transcription factor GATA-3 [107], the main regulator of IL-4/IL-5/IL-13 cytokine production in activated human CD4+ T cells [108]. PARP-1 has also a recognized role in the epigenetic control of gene expression by inducing chromatin remodeling through histone poly(ADP-ribosyl)ation (PARylation) and modification of the DNA methylation status [109]. Since epigenetic changes may promote Th2 cell differentiation by modulating IL-4 gene transcription [110], it cannot be ruled out that PARPi might favor a Th1 response also through this mechanism. Therefore, PARP inhibition-mediated effect on the fine-tuned balance underlying CD4+ Th differentiation within the TME represents an additional mechanism potentially improving the outcome of cancer immunotherapy. Nevertheless, PARP-1 inhibition has shown to modulate the differentiation of CD4+ T cells also by favoring the development of immunosuppressive regulatory T cells (Tregs). This effect is mediated by a decrease in the transcription factor FOXP3 destabilization, which is normally induced via PARylation [111][112][111,112], with consequent increased expression of genes controlled by FOXP3, such as those coding for CD25, CTLA-4 and interleukin 10 (IL-10). In mouse Tregs deficient for PARP-1, FOXP3 has been found at conserved non-coding DNA regions, a critical mechanism required for preserving FOXP3 gene expression in this cell subset [113]. Accordingly, treatment with the PARPi talazoparib has been correlated to increased Treg infiltration within the tumor site [98]. Conversely, notwithstanding a considerable up-regulation in terms of whole intra-tumor CD4+ T cell frequency, a remarkable change in immunosuppressive CD4+FOXP3+ Tregs was not observed in response to olaparib by Pantelidou et al. [87]. The authors suggested that further studies are required to establish the consequences of PARP blockade both in terms of overall impact on CD4+ T cell infiltration at the tumor site and of the role of different CD4+ T cell subgroups in the antitumor efficacy of PARPi. Moreover, in a preclinical in vivo model of SCLC, the tumor impairment observed with the combined use of PARPi and anti-PD-L1 was accompanied by reduced CD4+FOXP3 Treg frequency and amplified cytotoxic CD8+ T cell response [96]. Overall, the contrasting results reported suggest the requirement of further investigation to define the outcome of PARP inhibition on Treg development and function and the potential negative impact on ICI-mediated reactivation of T cell functionality in different cancer settings. Moreover, Treg induction mediated by PARPi can prevent the inflammatory response and the systemic toxicity potentially deriving from sustained STING pathway activation as well as by ICI co-administration. Remarkably, a direct crosstalk has been also identified between PARP-1 and PD-L1/PD-1 signaling pathway. Although not completely adequate for all tumor types, PD-L1 expression is still considered as one of the main biomarkers for predicting clinical response to anti-PD-1 mAbs [114]. Cumulative indications have suggested that PARPi can increase PD-L1 expression, likely through the activation of DNA damage-related STING and the downstream TBK1-IRF3-type I IFN signaling [115] or via the ATM-ATR-CHK1 pathway [116][117][116,117]. Moreover, inhibition of PARP-1, either by gene knockdown or pharmacological treatment with olaparib or talazoparib, has been shown to increase PD-L1 expression by deactivating the GSK-3β kinase, which promotes its proteasomal break-down [118][119][118,119], irrespective of the BRCA mutational status. Indeed, an increase in GSK3α/β phosphorylation at Ser21 and Ser9, which characterizes the inactive form of the kinase, has been observed [120]. As a result, despite being directly responsible for restraining T cell response, the increase in PD-L1 expression mediated by PARPi can amplify tumor sensitivity to anti PD-L1 mAbs. On the other hand, additional interference of PARPi with ICs-mediated signaling has been also demonstrated in preclinical mouse models of BRCA1-deficient ovarian tumors showing reduced expression of PD-1, Lymphocyte-activation gene 3 (LAG-3), and T-cell immunoglobulin (TIM-3), after combined PARPi and PD-1 blockade, resulting in reactivation of T cell functionality [98][121][122][123][98,121,122,123]. A potential drawback related to the increase in pro-inflammatory cytokines deriving from PARPi-mediated DNA damage could be represented by an enhancement of cancer initiation, promotion, and metastatic capacity [124]. In this context, a critical role may be played by the PARPi-mediated release of the inflammatory chemokine CCL5 in the TME [87]. In fact, in addition to favoring tumor infiltration of CD8+ T lymphocytes [87], the activation of the CCL5/CC type 5 chemokine receptor (CCR5) axis is involved in the development and progression of several malignancies, including breast and ovarian cancer, and in the generation of an immunosuppressive TME [125][126][127][128][125,126,127,128]. In particular, CCL5 has been shown to support the αvβ3 integrin-mediated tumor cell migration through activation of the PI3K/AKT and NF-kB signaling pathways [129], and by increasing the production of matrix metallopeptidases 2 (MMP-2) and MMP-9 [130][131][130,131], able to promote tumor cell invasiveness. Moreover, although being able to impair osteosarcoma growth, PARPi have been shown to support its metastatic potential through phosphorylation of the ezrin protein [132], a cytosolic structural molecule that regulates cell growth and motility [133]. This observation is of relevance because a high expression of the ezrin protein has been related to lower overall survival (OS) [134] and disease progression in patients with ovarian cancer [135], unveiling a potential mechanism of resistance to PARPi.2.2. PARP-1 Inhibition Favors NKG2D Activity
To provide the rationale for innovative combined therapeutic strategies effective in the context of diverse tumor immune landscapes, the identification of additional mechanisms underlying the immunostimulating effects of PARPi is strongly required. Type II transmembrane receptor Natural Killer Group 2 Member D (NKG2D) is a stimulatory receptor expressed by many immune cell subsets, including NK and CD8+ T cells, but also γδ and CD4+ T lymphocytes in definite settings, which promotes cytotoxic responses against target cells expressing its specific ligands. Although both human CD8+ T and NK cells express NKG2D [136][137][138][139][136,137,138,139], its role within these two distinct sub-populations is different. While in NK cells, the signaling pathway elicited by NKG2D is sufficient to induce killing of target cells [138], in CD8+ T cells, NKG2D delivers a co-stimulatory signal competent to support T cell receptor (TCR) activation [137][138][140][141][142][143][137,138,140,141,142,143], (although through distinctive pathways from those activated by the canonical CD28 co-stimulatory molecule) [144]. Of note, unlike CD28 which is represented within 50% of CD8+ T cells [145], NKG2D is ubiquitously expressed among this population, potentially representing an effective co-stimulatory alternative target in CD8+ T cells that lose CD28 following terminal differentiation driven by continuous stimulation [146]. Moreover, since the specific NKG2D ligands are more expressed than the CD28 counterparts CD80 and CD86, only present on APCs, NKG2D represents a valid co-receptor to be exploited in immunological approaches for cancer treatment. Distinctive NKG2D ligands include MHC-I-related chain (MIC) A/B and members of the ULBP family (UL16 binding proteins 1–6 in humans) [147]. In normal settings, NKG2D ligands are moderately expressed [148][149][148,149], but are promptly stimulated by stress conditions, including viral infection, cancer, DNA-damage, TLR signaling and cytokine-induced cell proliferation [140][147][148][140,147,148]. Although increased in stressed cells, NKG2D ligands are detected in normal tissues, including spleen, skeletal muscle, and skin [150]. Moreover, besides its co-stimulatory role, NKG2D is involved in the process of T cell memory progression. Of note, memory CD8+ T cells can be activated by cytokines under definite settings, which induce a specific NK-like killer phenotype, characterized by the acquisition of cytotoxicity towards tumor cells via NKG2D activation without requirement of TCR engagement [143][151][143,151]. The role of NKG2D in immunosurveillance against cancer has been demonstrated by data obtained in animal models showing that its deficiency leads to a reduced ability to fight tumors while the presence of its ligands has been shown to confer an effective barrier to cancer progression [140][148][140,148]. Although both primary tumors and metastases often accumulate MICA, MICB, and ULBP proteins [152], low or absent expression of NKG2D ligands represents a well-established strategy employed by leukemia cells for escaping the immune response [153][154][153,154]. Therefore, induction of NKG2D ligand expression may represent an appealing goal to be achieved to foster the impact of immunotherapies in cancer treatment. PARP-1 has been implicated in the negative control of the NKG2D ligands, MICA and MICB. Frequent relapse in acute myeloid leukemia (AML) has been ascribed to the persistence of leukemia stem cells (LSCs). These cells strongly contribute to drug resistance [155][156][155,156] and are characterized by poor expression of NKG2D ligands [157], which makes them distinctively less vulnerable as compared with bulk AML cells to the NKG2D-dependent effector functions. Of note, inhibition of PARP-1 has been shown to promote NKG2D ligands expression on LSCs, without affecting their expression on non-stem tumor or normal hematopoietic cells in a preclinical AML mouse model [158], providing a rationale to combine PARPi and NK cell-based immunotherapy for AML treatment [143]. CAR-T (chimeric antigen receptor T) cell therapies use genetically engineered T cells to contrast malignancies and have been proven beneficial in in patients with refractory B cell malignancies [159]. Most of the NKG2D-based CAR constructs are realized in T lymphocytes characterized by TCR heterodimers α and β, and comprise an extracellular, full-length human NKG2D linked to an intracellular CD3ς signaling domain [160]. Notably, the use of NKG2D-CAR T cells is currently being tested in the clinical practice, and data on NKG2D-based CAR clinical trials along with possible drawbacks have been recently provided [161]. However, expression of NKG2D ligands on normal cells, as a consequence of the activation of DNA repair mechanisms, may pose safety issues, due to the lethal toxicity possibly produced by off-tumor effects. In fact, preclinical mouse investigation has shown how adoptive transfer of NKG2D-CAR T cells can potentially promote fatal toxicities, although the severity of toxic effects was different depending on the mouse strain tested [162]. Overall, these observations suggest a possible use of PARPi in combination with NKG2D-based therapies, following careful consideration of the highly heterogeneous expression of NKG2D ligands either by cancer or normal cells. Key recognized modifications induced by PARPi in the TME are represented in Figure 1.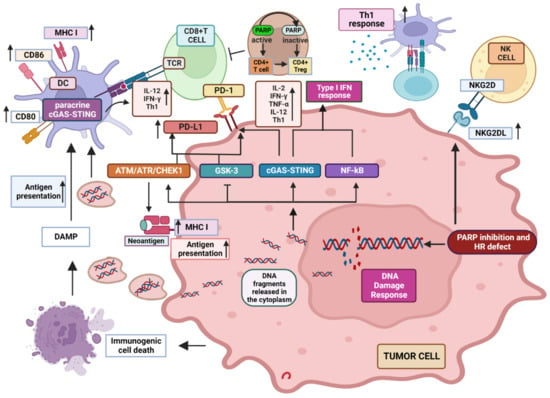
Figure 1. Recognized modifications induced by PARP inhibition in the TME. PARPi impair DNA repair which produces DNA fragments that are released in the cytoplasm, where they activate cGAS inducing STING pathway and the generation of a type I IFN inflammatory response. NF-kB is also activated, contributing to the production of inflammatory cytokines. DNA fragments can also be released extracellularly inducing paracrine GAS/STING response in DCs and increasing the expression of MHC molecules as well as antigen presentation. DDR response induces MHC expression in tumor cells, improving tumor-associated neo-antigen presentation induced by PARPi. PD-L1 expression is up-regulated via cGAS/STING, ATM/ATR pathway and GSK3 inactivation, potentially increasing the response to ICIs. PARPi induce NKG2D ligand expression, enhancing NK and CD8+ T cell anti-tumor cytotoxicity. See text for a more detailed explanation. “Created with BioRender.com.”