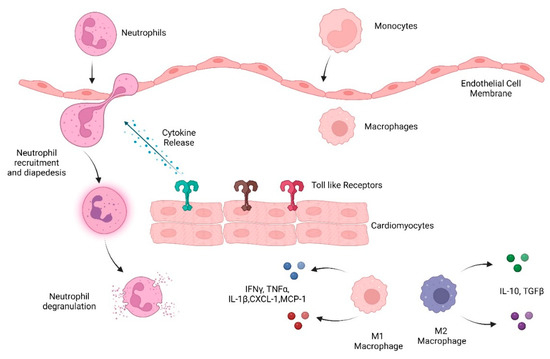
Innate immune cells are the early responders to infection and tissue damage. They play a critical role in the initiation and resolution of inflammation in response to insult as well as tissue repair. Following ischemic or non-ischemic cardiac injury, a strong inflammatory response plays a critical role in the removal of cell debris and tissue remodeling. However, persistent inflammation could be detrimental to the heart. Studies suggest that cardiac inflammation and tissue repair needs to be tightly regulated such that the timely resolution of the inflammation may prevent adverse cardiac damage. This involves the recognition of damage; activation and release of soluble mediators such as cytokines, chemokines, and proteases; and immune cells such as monocytes, macrophages, and neutrophils. This is important in the context of doxorubicin-induced cardiotoxicity as well. Doxorubicin (Dox) is an effective chemotherapy against multiple cancers but at the cost of cardiotoxicity. The innate immune system has emerged as a contributor to exacerbate the disease.
1. Introduction
Cardiovascular heart diseases (CVDs) are heart and vascular disorders and include coronary heart disease, cerebrovascular disease, peripheral arterial disease, rheumatic heart disease, and congenital heart disease. CVDs are the leading cause of morbidity and mortality globally
[1]. According to WHO, 17.9 million people died of CVDs in 2019, which represents 32% of global deaths. In the United States alone, about 697,000 deaths occurred due to CVD in 2020, which accounts for one in every five deaths
[2][3][2,3]. CVDs also represent a major economic burden on healthcare systems that costs over USD 300 billion each year
[4]. Studies have identified key molecular mechanisms leading to the development of therapeutic drugs. A growing body of evidence suggests that immune cells are key players in the development of CVDs and, in particular, inflammation triggers the early phases of the CVDs including the atherosclerotic process and myocardial infarction
[5][6][5,6]. Indeed, an increase in inflammatory cytokines is associated with an increased risk of developing CVDs. The CANTOS trial (Canakinumab Anti-inflammatory Thrombosis Outcome Study) demonstrated the key role of innate immune cells in CVDs
[7]. The study was the first to demonstrate that anti-inflammatory treatment by blocking pro-inflammatory IL-1β significantly reduced systemic inflammation and lowered the rate of recurrent cardiovascular events and cardiovascular death in patients with previous myocardial infarction (MI)
[7].
The anthracycline drug doxorubicin is a highly effective anti-cancer chemotherapy. Despite the fact that other anthracyclines have been developed, Doxorubicin (Dox) is widely prescribed to treat leukemia, lymphoma, Ewing sarcoma, osteosarcoma, neuroblastoma, and breast cancer
[8]. Studies suggest Dox intercalates DNA and disrupts topoisomerase-II-mediated DNA repair. Furthermore, it generates free radicals and induces damage to the cell membrane, DNA, and proteins
[9]. The clinical use of Dox is compromised by cardiac dysfunction that often progresses to heart failure. In contrast to the general population, young adult and childhood cancer survivors are at significant risk of developing cardiac failure
[8].
2. Inflammation in the Heart
Inflammation that occurs due to trauma or chemically induced injury and in the absence of pathogens is termed ‘sterile inflammation’. In the case of cardiac injury/damage, sterile inflammation forms the foundation of the first phase of cardiac remodeling and involves the production of chemokines and cytokines and the recruitment of innate immune cells such as neutrophils and macrophages
[10]. During tissue damage and inflammation, endogenous molecules related to injury are released as damage-associated molecular patterns (DAMPs). Innate immune cells recognize DAMPs via certain pattern-recognition receptors (PRRs) including Toll-like receptors (TLRs), nucleotide-binding and oligomerization domain (NOD)-like receptors, and C-type lectin receptors. Once these danger signals are detected, an inflammatory cascade is activated. This cascade includes the release of cytokines/chemokines, the activation of inflammatory pathways, and the subsequent recruitment of immune cells from circulation and bone marrow. The inflammatory phase promotes the clearance of necrotic cells and damaged tissues and is followed by a reparative phase of tissue repair that involves the deposition of a new extracellular matrix (ECM). This biphasic process is well regulated for efficient tissue repair. However, unchecked activation and chronic inflammation are detrimental and lead to aberrant cardiac damage.
Cardiac remodeling post ischemic injury occurs in three phases: inflammation, granulation, and maturation. The inflammatory phase is characterized by an increase in pro-inflammatory cytokines and chemokines, such as interleukin 1 (IL-1), IL-6, IL-8, tumor necrosis factor (TNFα), granulocyte colony-stimulating factor (G-CSF), GM-CSF, and CXCL-1. This leads to the recruitment of neutrophils and subsequently macrophages, two essential components of the myeloid system
[11]. The granulation phase involves ECM turnover and the differentiation of cardiac fibroblasts and the final maturation phase includes deposition of a new ECM including collagen. Excessive deposition of collagen, however, can lead to fibrosis, and subsequently heart failure. Cardiac fibrosis is a characteristic of several conditions, such as cardiomyopathy, MI, pressure overload, and the aging process
[12]. Below the researchers have discussed the role of the innate immune system in heart post damage (
Figure 1).
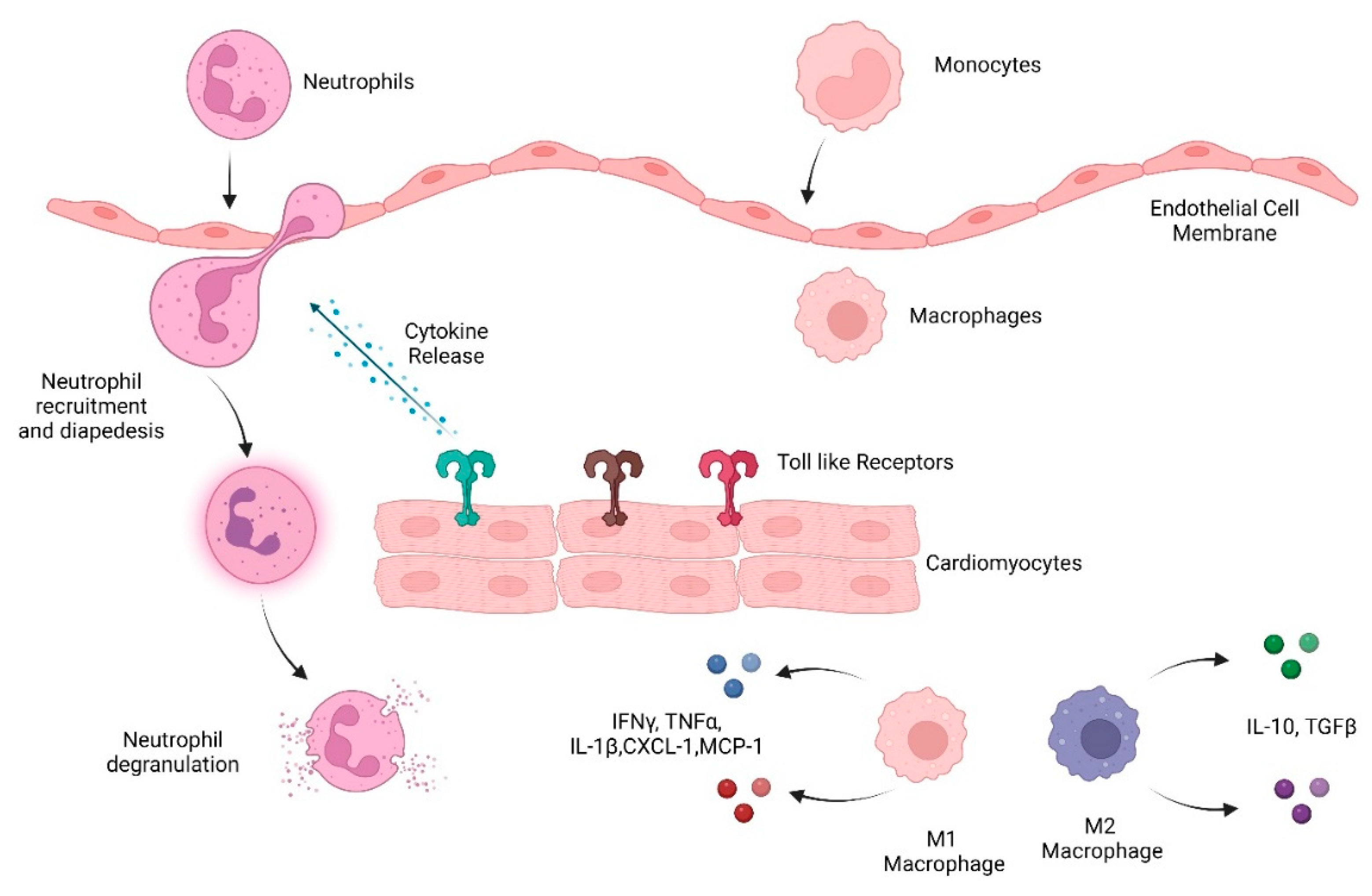
Figure 1. Overview of the innate immune system response to heart damage. Toll-like receptors detect damaged heart tissue and initiate an immune response including cytokine and chemokine release that recruits immune cells, including neutrophils, monocytes, and macrophages, to heart tissue.
3. Dox-Induced Cardiomyocyte Injury
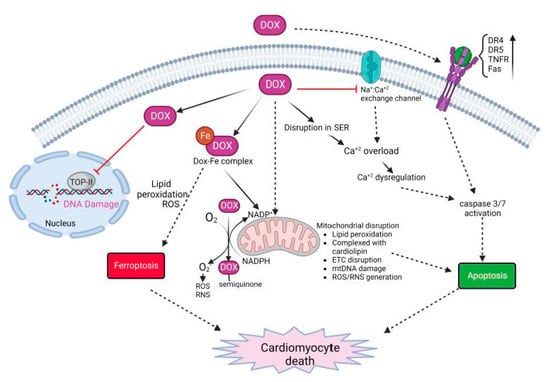
Dox-induced cardiomyocyte cell death is complex, and is regulated by multiple mechanisms of action (
Figure 2)
[13][14][15][62,63,64]. The major mechanisms leading to cardiomyocyte cell death include (1) the generation of reactive oxygen species (ROS) and nitrogen species (RNS) that leads to protein and DNA damage and lipid peroxidation; (2) the inhibition of topoisomerase-II (TOP-IIβ) resulting in DNA strand breaks; and (3) impaired mitochondrial function and the disruption of the electron transport chain (ETC). This leads to regulated or unregulated cell death apoptosis, ferroptosis and pyroptosis as well as necroptosis and the eventual release of inflammatory mediators
[16][65].
Figure 2. Molecular mechanisms on Dox-induced cardiomyocyte death. Dox treatment initiates multi-focal pathways including upregulation of death receptors, calcium overload, disruption in iron homeostasis, and generation of ROS/RNS. These events lead to oxidative stress, lipid peroxidation, DNA and protein damage, and cell death.
Several mechanisms lead to the generation and accumulation of ROS/RNS leading to oxidative stress in cardiomyocytes. Dox can directly bind to endothelial nitric oxide synthase (eNOS) and generate Dox-semiquinone radical. This generated superoxide (O
2−) radical. Furthermore, Dox increases the levels of inducible nitric oxide synthase (iNOS) and nitrotyrosine (NT). Superoxide and NT generate other potent oxidants that eventually cause apoptosis
[16][65]. In addition, cationic Dox can form a complex with free iron and form a Dox-Fe complex. The complex alters iron metabolism and directly interacts with free oxygen to generate ROS as well as enhances lipid peroxidation
[15][16][64,65]. This effect is enhanced by the upregulation of transferrin (TfR), allowing the transport of more iron into the cell leading to excess intracellular iron. This is furthermore complicated by the mitochondrial accumulation of iron in mitochondria. Dox downregulates ABCB8 protein, responsible for exporting iron outside the mitochondria, as well as inhibits mitochondrial ferritin thus disrupting the homeostasis of mitochondrial iron
[16][17][65,66]. The accumulation of Dox inside mitochondria is further enhanced by high-affinity binding to cardiolipin. Cardiolipin-bound Dox disrupts complexes I, III, and IV, which are essential in the electron transport chain (ETC) for generating ATP. Overall, the accumulation of Dox in mitochondria leads to enhanced ROS/RNS production, lipid peroxidation, DNA and protein damage, loss of ATP, and mitochondrial permeability
[14][63].
Dox also causes calcium dysregulation inside the cell, calcium overload, and induces apoptosis
[14][15][16][63,64,65]. Doxorubicinol, also known as DOXOL, is the hydroxyl metabolite of Dox that affects calcium homeostasis. DOXOL modulates calcium ATPase in sarco/endoplasmic reticulum (SER). It also disrupts the sodium/calcium exchange channel in SER, regulating the calcium level, which plays a critical role in cardiomyocyte contractility. Calcium overload activates calcium-dependent proteases such as calpains. Activated calpains cleave caspase-12 and induce the apoptosis of cardiac cells
[15][64]. Further, calcium overload activates calcium-dependent CaMKII (calmodulin activated protein kinase II) and PLN (phospholamban), which generate peroxides, activates caspase 3/9, and enhance apoptosis
[14][15][63,64].
Dox also induces cardiomyocyte apoptosis by both intrinsic and extrinsic mechanisms. Dox treatment causes oxidative stress by excess ROS/RNS, as described above, as well as mitochondrial damage. These events cumulatively cause the swelling of mitochondria, loss of membrane potential, and release of cytochrome c and apoptosis-inducing factor (AIF) in the cytosol. This leads to the activation of caspase 3/9 resulting in apoptosis and cell death
[14][16][63,65]. Further, oxidative stress activates HSF-1 (heat-shock factor-1) and stabilizes p53 protein. This leads to the upregulation of pro-apoptotic factors such as Bax, and downregulation of anti-apoptotic factors such as Bcl-XL
[15][64]. In addition, Dox also induces the upregulation of death receptors (DRs) on the cell surface. Death receptors such as TRAIL-receptor (tumor-necrosis-factor-related apoptosis-inducing ligand), Fas, DR4, DR5, and TNFR (tumor necrosis factor receptor), when bound to their cognate ligand, trigger the activation of caspase cascade ultimately leading to apoptosis
[14][16][63,65].
Another important cellular target of Dox is topoisomerase-II. Adult cardiomyocytes abundantly express Topoisomerase-IIß (TOP-IIß). TOP-IIß complexes with Dox and DNA to generate a ternary cleavage complex. The ternary complex induces single- and double-strand breaks in mitochondrial and nuclear DNA. DNA damage caused by Dox can lead to the overexpression of p53, leading to an increase in the expression of proapoptotic targets thus activating cell death. Furthermore, the ternary complex can lead to the inhibition of mitochondrial biogenesis and gene expression leading to the inhibition of secondary oxidative phosphorylation
[17][18][66,67].
3.1. Role of Immune Response in Dox-Induced Cardiotoxicity
Although studies have demonstrated the critical role of the innate immune system in the heart, the role of the innate immunity system in Dox-induced cardiotoxicity has not been assessed in great detail. The limited studies conducted in this field have identified some key components of the innate immune system that contributed to acute Dox-induced heart damage.
3.1.1. Cytokines/Chemokines
Several cytokines and chemokines have been targeted with respect to Dox-induced cardiotoxicity. In one study, IFN-γ was implicated in Dox-induced cardiotoxicity
[19][68]. IFN-γ was shown to reprogram lipid metabolism and sensitize cardiomyocytes to cardiotoxicity, which worsened heart function. Cardiomyocytes need fatty acids to develop respiratory capacity and impeding oxidation will interfere with that process. AMP-activated protein kinase (AMPK) signaling enhances fatty acid oxidation and helps regulate the respiratory capacity of cardiomyocytes. It was observed that, with Dox treatment, IFN-γ interfered with AMPK signaling by the suppression of the AMPK/ACC axis in a p38-dependent pathway, which enhanced the Dox-induced cardiotoxicity
[19][68]. Importantly, antibody treatment against IFN-γ improved the heart function in mice. This demonstrated that inhibiting IFN-γ could mitigate new as well as previously established Dox-induced cardiotoxicity. The investigators also found that IFN-γ inhibition had no effect on the therapeutic efficacy of Dox in mice with tumors
[20][69].
A study in breast cancer patients receiving Dox found that the plasma levels of cytokines CCL27 and macrophage migration inhibitory factor (MIF) were elevated after two cycles of Dox
[21][70]. CCL27 is associated with the homing of T lymphocytes to sites of inflammation whereas MIF is a crucial cytokine involved in acute and chronic inflammatory response. MIF has been found to play a role in maintaining cardiac homeostasis and found to be elevated in MI, atherosclerosis, and other disorders
[22][71]. MIF could play a role in protecting against cardiotoxicity by attenuating the loss of autophagy and ATP availability in the heart leading to the maintenance of cardiac homeostasis
[23][72]. CCL23, also called macrophage inflammatory protein 3, was also found to be elevated after each cycle. This cytokine has a suppressive effect on hematopoietic progenitor cells. Previous studies have shown an association between high levels of CCL23 and coronary atherosclerosis
[21][70]. Another study, conducted in HER2
+ breast cancer patients receiving anthracycline, revealed a significant increase in CXCL10 levels from baseline to post-anthracycline and post-trastuzumab treatment. This increase correlated with a decline in global longitudinal strain
[19][68]. CXCL10 has several roles, including serving as a chemoattractant for monocytes, macrophages, T cells, and NK cells and promoting T-cell adhesion to endothelial cells, thereby leading to the significant infiltration of these immune cells during cardiac remodeling
[24][73].
When aortas of mice treated with Doxorubicin were studied, there was a higher concentration of pro-inflammatory mediators such as IL-1β, IL-2, IL-6, and TNFα. TNFα levels had the highest elevation, which was associated with intrinsic wall stiffness that was prevented by the inhibition of TNFα
[25][74].
A study conducted in breast cancer patients who received Dox found that compared to baseline the levels of IL-10 were significantly increased 7 days after therapy completion in patients with cardiotoxicity. Increased levels of plasma NT-proB-type Natriuretic peptide (NT-proBNP, a marker for cardiac injury) correlated with the increased IL-10 levels in patients with cardiotoxicity. IL-10 levels were also positively correlated with IL-1β in the patients with cardiotoxicity, even though IL-10 is an immunosuppressive cytokine
[26][75].
3.1.2. TLRs
TLRs have also been found to be important as part of the innate immune response to Dox-induced cardiotoxicity
[27][76]. TLR5 was found to be significantly elevated in the hearts of mice treated with Dox. TLR5 deficiency led to reduced NADPH oxidase 2 (NOX2) levels in particular. NOX2 is an isoform of NADPH oxidase, a primary source of ROS in the heart. This was important as the investigators demonstrated that TLR5 activated NOX2 through Syk phosphorylation
[27][76]. TLR5 deficiency attenuated this effect. Dox was found to activate the p38 signaling pathway, which led to the apoptosis of cardiomyocytes. This p38 pathway was NOX2-dependent and hence activated by ROS. This pathway was also inhibited in TLR5-deficient mice
[27][28][76,77]. TLR5 deficiency led to lower TNFα and IL-1β mRNA levels and NF-κβ translocation was also inhibited in these mice and this led to improvements in heart function and less myofibrillar disruption in mice treated with Dox
[27][76].
In a study conducted in mice with TLR9 deficiency that was treated with Dox, it was found that cardiac function, myocardial fibrosis, and markers for myocardial damage were all reduced as compared to wild-type (WT) mice treated with Dox alone. TUNEL staining further revealed that in TLR9 KO mice with Dox treatment there was a significantly reduced number of apoptotic cardiomyocytes and reduced ROS production compared with wild-type (WT) mice with Dox treatment. Furthermore, it was found that TLR9 promoted the oxidative stress and apoptosis through p38 MAPK-dependent autophagy leading to the death of cardiac cells
[29][78].
Another TLR whose relationship to Dox-induced cardiotoxicity has been studied in mice is TLR2. TLR2-KO mice showed less NF-κβ activation, along with a lower production of pro-inflammatory cytokines (TNFα and IL-6), compared with WT mice. The TLR2-KO mice had higher survival rates than WT mice after Dox treatment. Furthermore, fewer TUNEL-positive cells were found in the myocardium, and caspase-3 activation was suppressed in the TLR2 KO mice with Dox treatment
[30][79]. In a study measuring inflammatory biomarkers in patients with heart failure, expression levels of TLR2 increased in patients in both the Dox group without heart failure and the Dox plus heart failure group
[31][80]. In another study evaluating the anti-inflammatory role of LCZ696 (sacubitril/valsartan), an angiotensin receptor neprilysin inhibitor that is used to reduce the risk of cardiovascular death for patients with heart failure, with respect to TLR2 deficiency, it was found that administration of the drug improved heart function and prevented cardiac fibrosis after Dox treatment. In addition, LCZ696 also prevented high TNFα expression. In TLR2-KO mice, similar results were observed, suggesting a connection between drug action and TLR2. Further studies found that LCZ696 attenuated the formation of the TLR2-MyD88 complex and this in turn alleviated the negative effects of Dox, as Dox promotes the formation of the TLR2-MyD88 complex
[32][81].
TLR4, a receptor of endotoxin, has also been shown to contribute to cardiac inflammation in Dox-induced cardiotoxicity. TLR4-KO mice had improved LV function and a reduction in cardiac ET-1, which contributes to heart failure. Additionally, when lipid peroxidation and nitrotyrosine were examined as markers of oxidative stress in TLR4-KO mice treated with Dox, there was significantly reduced oxidative stress. A study of an animal model of ischemia/reperfusion also suggested that TLR4 contributed to the development of oxidative stress. Furthermore, it was observed that the infiltration of lymphocytes, monocytes, and macrophages was reduced in the TLR4-KO mice treated with Dox compared to Dox-treated WT mice
[33][82]. Upregulation of the pro-apoptotic protein Bax was observed in WT Dox-treated mice, which was not seen in the TLR4-KO mice. These findings were confirmed by TUNEL assay where reduced apoptotic cells were seen in the TLR4-KO group. The study also found a significant upregulation of Bcl-2, an anti-apoptotic protein, in TLR4-KO mice with Dox treatment as compared to TLR4-KO mice. In a mouse study, downregulation of the GATA-4 pathway was seen in Dox-induced cardiomyopathy, and downregulation of this pathway is known to promote Dox-induced cardiotoxicity. However, in TLR4-KO mice, this downregulation did not occur, nor did the disease
[33][82]. Another finding that supports the importance of TLR4 in Dox-induced inflammation was that TLR4 expression was increased in macrophages following Dox treatment. When TLR4 was suppressed or depleted by injecting TAK-242 or using TLR4
lps-del mice, lower myofibrillar disruption as compared to Dox groups was observed
[34][83].
3.1.3. Innate Immune Cells
The role of innate immune cells, especially neutrophils, has been discussed in great detail in CVDs. However, the role of these immune cells in Dox-induced cardiotoxicity needs to be looked at in greater detail.
Neutrophils may also contribute to Dox-induced cardiotoxicity. In one study examining therapy-related clonal hematopoiesis following anti-tumor agents including Dox, cardiotoxicity was augmented by the infiltration and activation of neutrophils. In this study, an elevation of neutrophils was observed in cardiac tissue, which peaked at 7 days after treatment with a single bolus of Dox. When mice were transplanted with Trp53 heterozygous mutant bone marrow cells to establish a model of clonal hematopoiesis, neutrophil recruitment was higher in heart tissue compared to mice transplanted with WT cells post Dox treatment. Furthermore, when these heterozygous-Trp53-deficient neutrophils were analyzed for gene expression, these neutrophils were enriched for genes related to the inflammasome pathway (i.e.,
Nlrp1b, Gbp5, Il18) and chemokines (e.g.,
Ccl25, Ccrl2, and Cxcl1). When neutrophils were depleted in the mice with Trp53-deficient cells, an amelioration of echocardiographic parameters including fractional shortening was observed after Dox treatment indicating that neutrophil involvement is crucial for the detrimental effects of Dox
[35][84]. In another study conducted in breast cancer patients receiving anthracyclines, a high level of plasma neutrophil extracellular traps was seen to be associated with Dox-induced cardiotoxicity
[36][85].
Recently, the researchers demonstrated that Dox treatment induced a significant infiltration of neutrophils into hearts 24 h after Dox therapy, which was accompanied by an acute and late decrease in cardiac function, disruption in vascular structures such as pericytes and endothelial cells, and an increase in collagen deposition, leading to fibrosis. The depletion of neutrophils prevented Dox-induced cardiotoxicity with the preservation of vascular structures and prevention of excess collagen deposition 10 weeks after therapy
[37][86]. This effect was dependent on neutrophil elastase (NE) such that NE-KO mice treated with Dox had fewer apoptotic cardiomyocytes, preserved cardiac function, and preserved vascular structures compared to WT mice treated with Dox. Importantly, treating mice with a pharmacological inhibitor of NE (AZD9668) in conjunction with Dox significantly prevented the cardiotoxic effects of Dox. This study provided a potential therapeutic approach to mitigate the cardiac damage induced by Dox therapy. Additional studies are needed to elucidate the role of other neutrophil extracellular traps molecules in Dox-induced cardiac damage to more completely understand the role of innate immunity in the development of late cardiac morbidities in childhood cancer survivors
[37][86].
Dox induces sterile inflammation and non-ischemic cardiac damage characterized by systemic increases in TNFɑ, IL-1β, and LPS
[38][39][87,88]. The recognition of apoptotic cells, inflammatory mediators, and DAMPs by innate immune cells, macrophages, is critical for immune activation and resolution
[40][89]. Dox-treated apoptotic cells release TNF, which amplifies the inflammation in a TNF-R1-dependent manner. This was shown by significantly reduced levels of LDH, TNF, and neutrophils in TNF-R1-KO mice following doxorubicin administration
[38][87]. Interestingly, in a study conducted, it was shown that acute inflammation in response to doxorubicin is associated with the apoptosis of monocytes and macrophages. The investigators also found that the innate immune sensing of apoptotic cells is mediated by the TLR2/TLR9-MyD88 pathway and is key to the initiation of an acute inflammatory response to Dox
[39][88]. In addition to TLR2, TLR4 plays a role in cardiac inflammatory signaling, such that TLR4-KO mice treated with doxorubicin had improved LV function, reduced cardiac apoptosis, and reduced inflammatory mediators
[30][33][41][79,82,90]. As noted earlier, TLRs have been shown to play a major role in Dox-induced cardiotoxicity and activate innate immune cells, particularly macrophages, in response to injury and inflammation. Another study suggested a distinct role of TLR2 and TLR4 in mediating Dox-induced cardiotoxicity. An antibody-mediated blockade of TLR2 was associated with a reduced inflammatory response and attenuated Dox-induced cardiac dysfunction
[42][91]. This was in contrast to TLR4 blockage, where neutralizing antibodies to TLR4 exacerbated cardiac tissue damage. However, TLRs are also expressed by non-immune cells, including cardiomyocytes, and ligand stimulation induces cardiomyocyte apoptosis and inflammatory response
[43][44][92,93].
The understanding of the precise role of cardiac macrophages on the pathophysiology of Dox-induced cardiotoxicity is limited. Studies in mouse models of Dox-induced acute cardiotoxicity suggest that Dox treatment enhanced the M1 macrophage population and suppressed the M2 macrophage population
[45][46][47][45,50,51]. The M1 macrophage population has been shown to regulate the development of cardiac injury
[46][48][49][50,52,94]. In another study, the dynamics of circulating monocyte-derived recruited pro-inflammatory M1 macrophages and reparative anti-inflammatory M2 macrophages in doxorubicin-induced cardiotoxicity were shown to play a critical role in cardiac injury and repair
[50][95]. Using techniques, such as parabiosis, CX3C motif chemokine receptor 1 (CX3CR1)-based macrophage lineage tracing, and bone marrow transplantation, they showed that M1 macrophages may be the dominant population at the initial phase of cardiac injury, followed by a progressive increase in reparative M2 macrophages. This study suggests that resident macrophages are vulnerable to Dox insults but that the surviving resident macrophages are induced to proliferate
[50][95]. Importantly, the proliferation of the reparative M2 macrophages was dependent on scavenger receptor A1 (SR-A1), a critical regulator of DAMP-induced macrophage proliferation
[50][95]. In addition to pro-inflammatory mediators, M1 macrophages release pro-oxidative stress factors that sensitize cardiomyocytes to oxidative stress
[51][52][96,97]. Immunomodulatory approaches to regulate the balance of M1/M2 macrophage can help in the resolution of cardiac injury. Another study by Ye and colleagues demonstrated that IL-22 is a critical regulator of macrophage differentiation in response to cardiac injury
[52][97]. IL-22 deficiency reversed Dox-induced cardiac M1 /M2 macrophage imbalance and increased M2 macrophages. This effect was associated with reduced cardiomyocyte apoptosis, reduced cardiac vacuolization, and the improvement of cardiac function and LV tissue
[52][97]. Furthermore, in another study conducted by the same group, they demonstrated that Dox prevented M2 macrophage differentiation and that IL-12p35 deficiency exacerbated Dox-induced myocardial injury supporting the importance of M1 macrophages in Dox-induced cardiotoxicity
[53][98]. IL-12p35-KO promoted M1 macrophage differentiation, increased pro-inflammatory cytokines (IL-6, MCP-1, IL-1β, and IFN-γ), and reduced M2-macrophage-related anti-inflammatory cytokines (IL-4, IL-13 and IL-10)
[53][98]. Supporting the protective role of M2 macrophages, another study showed that glabridin, an isoflavone from licorice root, prevented Dox-induced cardiotoxicity by modulating Dox-induced gut dysbiosis and colonic macrophage polarization. Glabridin treatment reduced the level of LPS, increased butyrate, upregulated M2-related genes (arginase -1, CD206), and downregulated M1 genes (iNOS and CXCL9)
[54][99]. In a study conducted to examine the role of NLRP3, it was shown that NLRP3, despite involvement in sterile inflammation, regulates cardiac IL-10 production. NLRP3-deficient mice were susceptible to Dox-induced cardiomyopathy compared to WT mice. Mechanistically, NLRP3 regulates IL-10 production in cardiac reparative M2 macrophages, independent of IL-1ꞵ, and contributes to the pathophysiology of Dox-induced cardiotoxicity
[55][100]. As an immunomodulatory approach, Singla et al. showed that treatment with embryonic stem cells (ESCs) or exosomes derived from embryonic stem cells can promote M2 reparative macrophages and reduce inflammation-induced pyroptosis. This effect correlated with an increase in M2 macrophages and improved cardiac function
[56][101]. Furthermore, Liu et al. showed that the adoptive transfer of bone-marrow-derived M2 macrophages prevented Dox-induced cardiac remodeling and injury. M2 macrophage transplantation transferred mitochondria to cardiomyocytes, alleviated doxorubicin-induced cellular stress, and reduced cardiac apoptosis
[57][102].
Invariant natural killer T-cells (iNKT) are a subset of T-lymphocytes that express properties of both T cells and natural killer cells. Studies have shown that iNKT cells modulate cardiac tissue inflammation. For example, treatment with alpha-galactosylceramide (αGC), an activator of iNKT cells, prevented damage after MI. Another study looked at these cells in the context of Dox-induced cardiotoxicity and found that heart function was normal in mice treated with Dox and αGC simultaneously but not in mice treated with Dox only. Additionally, an analysis for fibrosis found that there was low collagen deposition in the mice treated with Dox+ αGC compared to mice with treated with Dox only. A qPCR analysis revealed that M2 macrophage expression was higher in mice treated with Dox+ αGC mice than in mice with Dox only. In this study, it was concluded that iNKT cell activation prevented Dox-induced LV dysfunction and cardiac fibrosis in mouse hearts hinting at a role for these cells in Dox-induced cardiotoxicity
[58][103].