1. Asymptomatic Hyperuricemia in CKD
A systematic review and meta-analysis recently reported that higher uric acid levels increased the risk of CKD incidence and progression [
17]. However, whether to treat or not to treat asymptomatic hyperuricemia in CKD patients is still under debate. Two questions need to be answered regarding the pathogenic role of uric acid in CKD. First, does hyperuricemia accelerate the progression of CKD? Two recent randomized controlled clinical trials reported negative results for this question [
18]. In the CKD-FIX trial, 363 patients with stage 3 or 4 CKD and no history of gout were randomized to receive allopurinol (100–300 mg/day) or placebo for approximately 1.5 years. Despite the lowering of serum uric acid from 8.2 mg/dL to 5.1 mg/dL, allopurinol treatment did not significantly slow a decline in eGFR compared to the placebo [
19]. The PERL trial studied 530 patients with type 1 diabetes, evidence of kidney disease (eGFR, 40–100 mL/min/1.73 m
2), and serum uric acid levels > 4.5 mg/dL. Participants received either allopurinol (100–400 mg/day) or placebo for 3 years. Despite the lowering of serum uric acid from 6.1 mg/dL to 3.7 mg/dL, the average annual loss in measured GFR did not differ between the two groups [
20]. Although these studies had high dropout rates (20–30%), the results were compatible with a Mendelian randomization study [
21], suggesting that uric acid does not predict CKD progression. The outcomes might have been different if the mean baseline uric acid levels had been higher, e.g., approximately 10 mg/dL.
Interestingly, febuxostat might have some favorable effects on renal progression. Febuxostat slowed a decline in eGFR over 6 months in 45 patients with CKD stages 3 and 4, compared to 48 in the placebo group [
22]. In cardiac surgery patients with an eGFR ≤ 60 mL/min/1.73 m
2, kidney function was more preserved by febuxostat than by allopurinol [
23]. Larger clinical trials are necessary to compare the effects between febuxostat and allopurinol.
The next question to consider is whether hyperuricemia increases the incidence of CKD. Observational studies consistently report that hyperuricemia predicts the development of CKD [
24]. However, the pathogenic role of uric acid in new-onset CKD is unclear. Hassan et al. recently examined the association between uric-acid-lowering therapy and the incidence of CKD in a large cohort of U.S. veterans with no pre-existing CKD and found that, in patients with kidney function within the reference range eGFR
> 60 mL/min/1.73 m
2, uric-acid-lowering therapy was not associated with the preservation of kidney function. In patients with baseline serum uric acid levels of ≤8 mg/dL, uric-acid-lowering therapy was paradoxically associated with a higher risk of incident CKD, indicated by the development of an eGFR < 60 mL/min/1.73 m
2 or new-onset albuminuria [
25]. Therefore, evidence for the causal effect of uric acid on CKD is currently lacking. Properly powered randomized clinical trials in patients with no pre-existing CKD are necessary.
2. Diabetic Kidney Disease (DKD)
Hyperuricemia is often associated with diabetes mellitus (DM) [
11], and it seems that insulin resistance correlates with serum uric acid levels and inversely correlates with the renal clearance of uric acid [
26]. In addition, hyperuricemia may be associated with an increased risk of DKD in patients with type 2 DM [
27]. However, the causal relationship between hyperuricemia and DM or DKD has not been documented.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are first-line therapeutics in DKD because of their cardiorenal protection. They have a uric-acid–lowering effect, which was confirmed by a meta-analysis of 62 clinical trials [
28]. Treatment with an SGLT2 inhibitor consistently lowered serum uric acid concentrations in a total of 34,941 patients with type 2 DM. When baseline uric acid levels were within the normal limit, the SGLT2 inhibitors typically decreased serum uric acid concentrations by 0.60–0.75 mg/dL in trials lasting 6–12 months. This uric acid–lowering effect was rapidly induced within days and persisted throughout trials of a 2-year duration [
29].
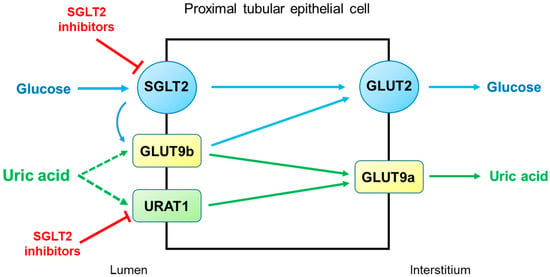
SGLT2 inhibitors reduce serum uric acid concentrations by elevating renal uric acid excretion. However, the molecular mechanisms by which SGLT2 inhibitors exert uricosuric action are unclear. It seems likely that SGLT2 inhibitors induce hyperuricosuria by reducing reabsorption rather than by enhancing the secretion of uric acid in the proximal tubule (
Figure 3). As described above, URAT1 and GLUT9 are the major uric acid transporters for reabsorption. GLUT9 has two isoforms, GLUT9a and GLUT9b, which transport both glucose and uric acid in the proximal tubule [
30]. Unlike GLUT9a, GLUT9b is presumed to be located in the apical membrane and is overwhelmed by excessive glucosuria when SGLT2 is inhibited, with its capacity to reabsorb uric acid markedly diminished [
29]. On the other hand, the role of URAT1 in the uricosuric effect of SGLT2 inhibition was proposed in mice [
31]. This was also suggested in humans because uricosuria induced by combination therapy with empagliflozin and benzbromarone did not differ from benzbromarone monotherapy [
32]. Thus, URAT1 seems to be the target of SGLT2 inhibitors to induce hyperuricosuria.
Figure 3. Potential mechanisms by which SGLT2 inhibitors increase uricosuria in the proximal tubule. URAT1 and GLUT9 are the major pathways for uric acid reabsorption. When SGLT2 is inhibited by SGLT2 inhibitors, GLUT9b is overwhelmed by excessive glucose and its capacity for uric acid transport is diminished. SGLT2 inhibitors can also inhibit URAT1, further increasing uricosuria. Abbreviations: GLUT9, glucose transporter 9; SGLT2, sodium-glucose cotransporter 2; URAT1, urate transporter 1.
3. ADTKD
ADTKD is a rare genetic kidney disease characterized by tubular damage and interstitial fibrosis that can progress to end-stage kidney disease. More accurately, ADTKD is a group of conditions with autosomal dominant inheritance. Mutations in UMOD and MUC1 are the typical causes of ADTKD, but other rarer (REN, SEC61A1), atypical (DNAJB11), or heterogeneous (HNF1B) subtypes have also been described [
33]. UMOD encodes uromodulin, the most abundant protein secreted in normal urine, but with unidentified multiple roles in renal physiology and pathophysiology. MUC1 encodes transmembrane epithelial mucin-1 and has roles in epithelial barrier protection. REN encodes preprorenin, which is converted to prorenin and renin, and has a role in regulating blood pressure and electrolyte balance. HNF1B encodes homeodomain-containing transcription-factor hepatocyte nuclear factor 1β (HNF1β), which has a role in the development of several organs, including the kidneys. Because of increased awareness and genetic testing, ADTKD has become the third-most-common hereditary monogenic kidney disease in Western countries, after autosomal dominant polycystic kidney disease and type IV collagen mutations [
34]. Until its recent definition as a distinct disease entity, the following various terms were used to describe this disorder: uromodulin kidney disease, uromodulin-associated kidney disease, mucin-1 kidney disease, familial juvenile hyperuricemic nephropathy, medullary cystic kidney disease type 1, medullary cystic kidney disease type 2, and renin-associated kidney disease. Because these terms were confusing or misleading, an international consensus by Kidney Disease Improving Global Outcomes (KDIGO) was reached to use the term “autosomal dominant tubulointerstitial kidney disease”, with a subclassification based on underlying genetic defects [
34].
Typical ADTKD patients can present with asymptomatic azotemia incidentally found during routine laboratory testing [
35]. This diagnosis should be suspected when slowly progressive CKD is accompanied by an absence of significant proteinuria with bland urine sediment, normal or slightly elevated blood pressure, normal- or small-sized kidneys, and a positive family history of progressive CKD [
33,
34]. A precise diagnosis can be made genetically, and a kidney biopsy is usually unnecessary because it shows non-specific interstitial fibrosis and tubular atrophy [
35].
Notably, hyperuricemia and secondary gout are important clinical clues for the diagnosis of ADTKD. In patients with ADTKD-UMOD and ADTKD-REN, hyperuricemia frequently occurs in childhood, and early-onset gout can present during the teenage years with hypouricosuric hyperuricemia [
34,
36]. Although the molecular mechanisms by which hyperuricemia is induced are unclearly defined, proximal tubular adaptations are conceivable in response to plasma volume contractions induced by salt-losing nephropathy in ADTKD-UMOD and anemia and hypotension in ADTKD-REN. A compensatory response in the proximal tubule may accompany the enhanced reabsorption of uric acid and lead to hyperuricemia [
37]. Aged uromodulin knockout mice, in addition to hyperuricemia and hypertension, had an upregulation of uric acid transporter URAT1 and sodium-hydrogen exchanger 3 (NHE3) in the proximal tubule [
38].
In contrast, ADTKD-MUC1 has a prevalence of hyperuricemia and gout similar to those of other advanced kidney diseases [
39]. As in other cases of CKD, allopurinol and febuxostat are indicated to lower serum uric acid levels. Hyperuricemia and hypomagnesemia are characteristic laboratory features in ADTKD-HNF1B. In particular, hyperuricemia was reported in 37% of pediatric patients with an early onset at infancy [
40] and in 20% of adult patients [
41]. The reason why hyperuricemia is more frequently reported in infants needs to be explained. Renal magnesium wasting in ADTKD-HNF1B may be related to the finding that transcription of FXYD2, the γ-subunit of the Na
+/K
+-ATPase, is regulated by HNF1β in the distal tubule [
42]. In the distal convoluted tubule, transcellular Mg
2+ uptake is driven by Na
+/K
+-ATPase activity, which controls the membrane potential at the apical membrane [
34].
4. Thiazide and Loop Diuretics
Hyperuricemia is frequently complicated by diuretic use, and thiazide and loop diuretics are the major causes of secondary hyperuricemia [
43]. Diuretic-induced hyperuricemia may occur within a few days after the initiation of medication, appears to be dose-dependent, and persists during the period of administration [
43,
44].
Thiazide and loop diuretics can directly and indirectly increase the reabsorption of uric acid in the proximal tubule. The direct interaction of diuretics with uric acid transporters for reabsorption or secretion may occur, and diuretic-induced volume depletion indirectly increases uric acid reabsorption by the proximal tubule [
45].
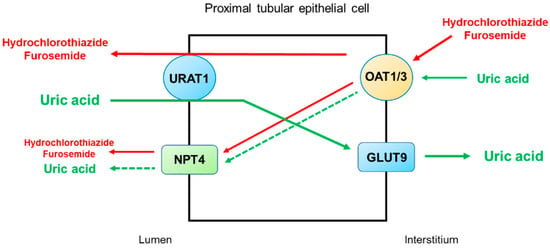
Figure 4 illustrates the secretory pathways of thiazide and loop diuretics for delivery to the tubular sites of action and the potential inhibition or stimulation of uric acid transporters in the proximal tubule. Thiazide and loop diuretics enter the proximal tubular cell via basolaterally located organic anion transporters OAT1 and OAT3, and they may exit via apically located URAT1 or NPT4 [
46]. On the other hand, URAT1 is the major transporter for uric acid reabsorption, and OAT1 and NPT4 are the major pathways for uric acid secretion. Therefore, competitive binding between diuretics and uric acid for the basolateral OAT1 and OAT3 and apical NPT4 would reduce uric acid secretion by the proximal tubule. The apically located URAT1 acts as an anion exchanger and may reabsorb uric acid in exchange for the secretion of thiazide or loop diuretics [
47]. Thus, a reduction in uric acid secretion, as well as an increase in uric acid reabsorption by the proximal tubule, will lead to diuretic-induced hyperuricemia.
Figure 4. Potential mechanisms by which thiazide and loop diuretics reduce uricosuria in the proximal tubule. URAT1 is the major transporter for uric acid reabsorption, and OAT1 and NPT4 are the major pathways for uric acid secretion. Thiazide and loop diuretics enter the proximal tubular cell basolaterally via OAT1 and OAT3 and exit via apically located URAT1 or NPT4. Competitive binding between diuretics and uric acid for the basolateral OAT1 and OAT3 and apical NPT4 would reduce uric acid secretion by the proximal tubule. The apically located URAT1 acts as an anion exchanger and may reabsorb uric acid in exchange for the secretion of thiazide or loop diuretics. Thus, a decrease in uric acid secretion, as well as an increase in uric acid reabsorption by the proximal tubule, will lead to diuretic-induced hyperuricemia. Abbreviations: GLUT9, glucose transporter 9; NPT4, sodium-dependent, inorganic phosphate transporter 4; OAT1, organic anion transporter 1; OAT3, organic anion transporter 3; URAT1, urate transporter 1.