Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zeyong Yang and Version 2 by Conner Chen.
Advances in molecular biology technology have piqued tremendous interest in glycometabolism and bioenergetics in homeostasis and neural development linked to ageing and age-related diseases. Methylglyoxal (MGO) is a by-product of glycolysis, and it can covalently modify proteins, nucleic acids, and lipids, leading to cell growth inhibition and, eventually, cell death. MGO can alter intracellular calcium homeostasis, which is a major cell-permeant precursor to advanced glycation end-products (AGEs). As side-products or signalling molecules, MGO is involved in several pathologies, including ageing.
- methylglyoxal
- glyoxalase
- neurodegenerative
- bioenergetics
1. Methylglyoxal (A Metabolic Side-Product) and the GLO System In Vivo
Methylglyoxal (MGO) is an unavoidable endogenous coproduct of the metabolism of carbohydrates, lipids, and proteins that is produced either autonomously or enzymatically [1] primarily via nonenzymatic degradation of the triphosphate intermediates of glycolysis.
The glyoxalase (GLO) system primarily catalyses the detoxification of MGO and other reactive aldehydes. In the glutathione (GSH)-dependent GLO1 system, MGO is transformed into S-D-lactoylglutathione and then into D-lactate via the GLO2 system. Concurrently, GLO1 activity limits the reaction rate of MGO degradation and regulates MGO-induced toxicity. Many of the cellular signalling pathways that regulate GLO1 prevent the formation of advanced glycation end-products (AGEs) during MGO anabolism or catabolism [2].
It is of paramount importance to thoroughly study antiapoptotic GLO2 overexpression [3]. Transduced recombinant Tat-GLO1, Tat-GLO2, or both have been studied, and these are believed to defend HT22 cells against the toxicity of MGO and H2O Indeed, Tat-GLO1 and Tat-GLO2 have been shown to provide toxicity protection in vivo in diabetic mouse models with ischaemic damage [4]. Interestingly, a substantial increase in intracellular Tat-GLO2 has a protective effect on HT22 cells but to a lesser degree than a similar increase in Tat-GLO The activities of GLO1 or GLO2 can increase pyruvaldehyde elimination. Under normal conditions, cells are not affected by MGO-induced toxicity, and the GLO system is the most pivotal signal pathway for the detoxication of this substance.
However, more experimental strategies are necessary. Liquid chromatography–mass spectrometry (LC-MS/MS) of glycation adducts is an excellent technique for comparing various AGE sources; however, this method does not provide relevant data on homologous proteins. Nonetheless, because both the formation and degradation of glycation products depend on the protein sequence and structure to some extent [5], this information is needed to study functional changes in glycation-related proteins.
The cerebral GLO system can adapt adequately to eliminate MGO-induced hazardous substances such as side-products while still supplying sufficient energy for brain functioning (Figure 1).
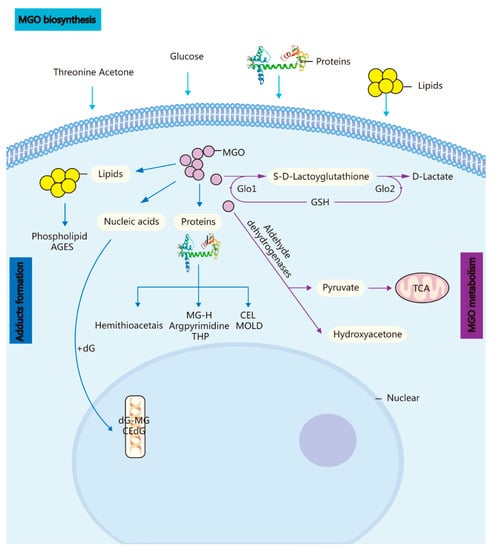
Figure 1. Biosynthesis, metabolism, and adduct formation in MGO. MGO biosynthesis: MGO is an unavoidable endogenous coproduct of the metabolism of glucose, threonine acetone, lipids, and proteins. Adduct formation: The incubation of these highly reactive compounds with proteins, lipids, and nucleic acids leads to the rapid formation of AGEs and advanced lipoxidation end-products. MGO metabolism: MGO is converted into S-D-lactoylglutathione in the GSH-dependent GLO1 system and subsequently transformed into D-lactate via GLO Methylglyoxal reductase and aldehyde dehydrogenase convert MGO into hydroxyacetone or pyruvate.
2. Potential Role of MGO in Ageing via Its Induction of Oxidative Stress, Neuropathic Pain, an Anxiolytic Effect, and Apoptosis [6][7]
High MGO levels can render neurons susceptible to AGE formation, a risk factor linked to the development of neurodegenerative disorders [7][9]. MGO can induce oxidative stress and cause irreversible loss of protein function, including protein cross-linking.
MGO accumulation has been implicated in multiple neurodegenerative diseases, and AGEs in combination with MGO can potentially induce ageing and diabetes-associated complications. Occurrences of these metabolic adaptations of neural cells have been found in Drosophila [8][10] and mice [9][11]. The key role of glycolytic-derived lactate released from astrocytes during neural activities has been identified [10][12]: this lactate acts as an intercellular chemical messenger [11][13] and metabolic fuel [12][14]. By observing the brain mitochondria bioenergetics and oxidative status of rats that have undergone chronic treatment with MGO and/or pyridoxamine (a glycation inhibitor), it has been found that the resulting increased MGO levels induce mitochondrial impairment in the central nervous system (CNS) and that pyridoxamine cannot reverse MGO-mediated effects.
Increased intracellular MGO may induce cytotoxicity in INS-1 cells. This occurs primarily via the activation of oxidative stress and is further amplified by its triggering of the mitochondrial apoptotic pathway and the endoplasmic reticulum (ER) stress-evoked JNK pathway, which are involved in glucotoxicity-mediated pancreatic beta-cell apoptosis. Furthermore, the cytotoxicity of MGO is mediated via the regulation of the amount of deoxyribonucleic acid and the activation of apoptosis. In the development of diabetic complications, MGO can modify cellular proteins (i.e., cross-linking amino groups), which then generate AGEs. Numerous studies have highlighted the causal relationship between MGO-derived AGEs and diabetic complications and the role of AGEs in endothelial dysfunction [13][15].
It has been demonstrated that AGEs can induce corneal endothelial cell apoptosis by increasing cellular oxidative stress [14][16]. MGO-induced reactive oxygen species (ROS) and AGEs can impair mitochondrial function, resulting in further ROS production and damage. AGE accumulation increases significantly after MGO treatment and can be alleviated via treatment with Tan IIA. Furthermore, Tan IIA treatment also attenuates the production of ROS, TBARS, and H2O2 in cultured human brain microvessel endothelial cells (HBMECs). These findings indicate that Tan IIA can protect HBMECs by inhibiting AGE accumulation and reducing oxidative stress.
In addition, MGO requires between several hours and days to induce apoptosis. Furthermore, the effects that MGO has on GABAA receptors occur only at lower concentrations and are momentarily dissociated because of their more prominent effects, such as cytotoxicity and AGE formation. Diabetes, which has been linked to stroke, can elevate blood MGO levels via an increase in MGO secretion due to high glucose levels, thus impairing the GSH-GLO system, which accelerates MGO elimination. MGO also glycates proteins and causes dicarbonyl stress. This is evident in the diabetic brain, in which there is only a tiny chance of GSH-dependent MGO elimination, resulting in elevated protein glycation and oxidative or carbonyl stress. Furthermore, these MGO-induced changes may exacerbate post-stroke brain damage. Administering N-acetylcysteine (NAC) may alleviate post-stroke brain injury damage by restoring GSH generation and normalising the MGO-to-GSH ratio, thus diminishing oxidative or carbonyl stress levels. This treatment may be a critical factor in the management of diabetic stroke risk and outcomes.
There may be a metabolic imbalance following acute neuronal lesions. An increase in the rate of glycolysis is accompanied by MGO formation, leading to metabolic dysfunction and inflammation. As mentioned earlier, the GLO system is the primary MGO detoxification system; however, it can be impaired following excitotoxicity and stroke. Changes in GLO1 protein content are associated with excitotoxicity but seemingly not with fibre transection. Cell-specific changes in GLO1 immunoreactivity following different in vitro and in vivo lesion types may be a common phenomenon in the aftermath of neuronal lesions [13][15]. Furthermore, MGO triggers protein and nucleotide modification (e.g., AGEs), ROS production, and apoptosis. MGO has been shown to increase the production of ROS in other animal disease models [14][16]. In addition to directly increasing ROS production, MGO can also increase oxidative stress by inducing AGE formation. Specifically, MGO modifies proteins and DNA to form hydroimidazolone MGO-H1 and imidazopurinone MGOdG adducts, respectively. An abnormal accumulation of MGO and dicarbonyl stress increases adduct levels, which may induce apoptosis and replication catastrophe [15][17].
There is mounting evidence supporting the role of GLO1 and its substrate (MGO) in the regulation of anxiety-like behaviour [16][17][18][18,19,20]. Changes in the brain expression levels of GLO1 and Gsr produce a significant impact on anxiety-related behaviours, thus establishing the causal role of these genes. GLO1 plays an essential role in MGO clearance via the excessive secretion of GLO1, preventing MGO accumulation and inhibition of GLO1, resulting in MGO aggregation [16][17][18][18,19,20]. Evidence has been presented showing that MGO can reduce anxiety-like behaviour in an elevated plus maze. The research findings referenced here are based on the results of administering MGO to the basolateral amygdala (BLA), which is closely linked with anxiety-like behaviour [19][20][21][22][21,22,23,24]. Furthermore, astrocytes and neurons have different energy-adaptive mechanisms for GLO defence mechanisms, which may function as a shield mechanism, preventing MGO from causing cellular injury. MGO increases intracellular calcium in sensory neurons and causes behavioural injury via the transient receptor potential ankyrin 1 (TRPA1) channel. In addition, after the administration of MGO in wild-type rats and mice, the immunohistochemical phosphorylation of extracellular signal-regulated kinase (p-ERK) and multiple pain-like behaviours were evaluated. It was observed that MGO stimulated certain behaviours, including conditioned place avoidance (a measure of affective pain), dose-dependent licking, enhancement of injury perception, heat hypersensitivity, and mechanical stimulation.
MGO-mediated dicarbonyl adducts have complex pleiotropic effects on biological processes in cells. These adducts can modulate protein activity and stability and generate ROS and oxidative stress [23][25], which may induce distinct outcomes [23][25]. They have been shown to contribute to oxidative DNA damage and apoptosis. DNA glycation is associated with elevated mutation frequency, DNA strand breaks, and cytotoxicity [24][26]. Parkinsonism-associated protein DJ-1 is a major nucleotide repair system. Furthermore, some MGO-derived AGEs possess antioxidant properties [25][27].
GLO1- and MGO-derived AGEs play major roles in vascular physiology and pathophysiology, including the etiopathogenesis of brain microvascular endothelial barrier dysfunctions [26][27][28,29]. High concentration levels of MGO can induce mitochondrial injury, ROS generation, and oxidative-stress-mediated cell damage [28][30]. In addition, MGO toxicity within cells or tissues is mediated primarily via an enhancement of oxidative stress and apoptosis abduction. In light of these processes, oxidative stress is considered to play a crucial role in the pathogenesis of Alzheimer’s disease (AD) [29][30][31,32]. A marked increase (less than three-fold) took place in AMPK phosphorylation at 3, 6, and 18 h after 5 mM MGO treatment, indicating that autophagy was induced through AMPK activation. This strong AMPK activation was confirmed by enhanced phosphorylation of the AMPK substrate. AMPK phosphorylates and activates the tuberous sclerosis complex and then phosphorylates the small GTPase Ras homolog enriched in the brain. This is done to prevent mTOR activation. DNA glycation is linked to elevated mutation frequency, DNA strand breaks, and cytotoxicity. Parkinsonism-associated protein DJ-1 is a major nucleotide repair system [24][26] (Figure 2).
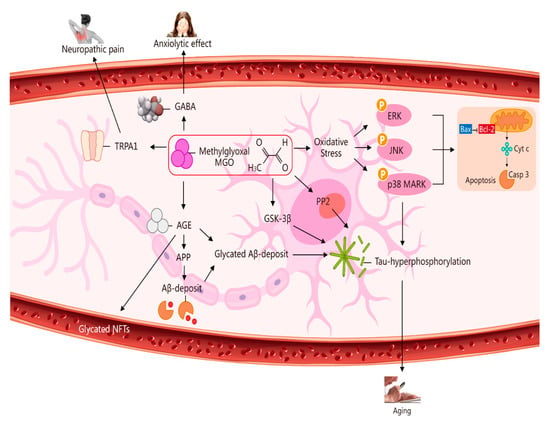
Figure 2. Multiple pathways via which MGO induces oxidative stress, pain, anxiolytic effects, and ageing. The pathways activated by MGO induce oxidative stress, which may play a role in ageing. The formation of AGEs involves nonenzymatic reactions that occur when sugars or dicarbonyl compounds, such as MGO, are reduced. Research has been conducted on the connection between neuropathic pain and the transient receptor potential ankyrin 1 (TRPA1) pathway. MGO mediates the anxiolytic effect via the modulation of GABAergic actions. MGO mediates ERK-, JNK-, and p38-dependent endothelial cell inflammatory responses, which may occur independently of oxidative stress. In addition, BCL2-associated X protein (BAX) accesses mitochondria to induce cytochrome c release, and Bcl-2 inhibits BAX translocation from cytosol to mitochondria during relevant apoptosis. MGO induces the expression of phosphorylated tau protein and increases the expression of protein phosphatase 2A (PP2A) and glycogen synthase kinase-3β (GSK-3β). A reduction in Aβ deposition can eventually improve learning and memory ability. AGE interaction with the receptor for AGEs (RAGE) carries an implication for AD. Glycated Aβ (Aβ-AGE) can aggravate an AD-like pathology via the receptor for the RAGE pathway. MGO increases intracellular calcium in sensory neurons and produces neuropathic pain via the cation TRPA1 channel.