Adenosine deaminase (ADA, EC 3.5.4.4) - the enzyme engaged in purine metabolism that irreversibly converts adenosine or 2′deoxyadenosine to inosine or 2′deoxyinosine, respectively. In human tissues, it occurs as two isoenzymes: ADA1 and ADA2. ADA1 constitutes the majority of ADA activity and it is present in virtually all tissues, while ADA2 has been found with ADA1 only in monocytes/macrophages. Both ADA isoenzymes are present in cytosolic form, soluble fraction that can be located away from the originating cell, or as ecto-enzymes binding to the cell surface by dedicated proteins. Intracellularly, ADA plays a significant role counteracting high concentrations of 2'deoxyadenosine. When intracellular ADA activity is lowered, e.g. during severe combined immunodeficiency disease (SCID), 2′deoxyadenosine accumulates and is converted to 2′-deoxyadenosine-5′-triphosphate (dATP), which inhibits ribonucleotide reductase (RNR), a crucial enzyme in DNA synthesis that follows disrupted T-cell development. Intracellular concentration of adenosine, the second substrate for ADA, is maintained inside the cell on the low level by adenosine kinase with Km value ~ 1 µM. Under conditions of low energy charge, adenosine-5′-monophosphate (AMP) that originates from adenosine-5′-triphosphate (ATP) degradation, is rapidly transformed to adenosine, which is not immediately deaminated to inosine due to high Km of ADA (25–100 µM). This results in temporary accumulation of adenosine that is vigorously exported out the cell via equilibrative nucleoside transporters (ENTs). Therefore, transmembrane adenosine transport, together with the activities of ecto-ADA and soluble ADA are important regulators of extracellular adenosine concentration. Except enzymatic function, ecto-ADA plays a significant extra-enzymatic role in the interactions between cells that expressed ADA-anchoring proteins on their surfaces. This co-stimulatory and cell-to-cell connecting actions along with its activity regulate many cellular processes related to proliferation and differentiation, which affect pathological conditions associated with cardiovascular diseases such as endothelial activation and dysfunction, inflammation, myocardial ischemia-reperfusion injury, or coagulation disorders. Since these pathologies are associated with ADA overexpression, the inhibition of its activity as well as binding to the surface proteins exhibit an attractive therapeutic potential in cardiovascular diseases.
- adenosine deaminase
- adenosine
- adenosine deaminase 1
- adenosine deaminase 2
- cardiovascular diseases
- inhibition
- deoxycoformycin
- pentostatin
- EHNA
1. Introduction
Adenosine deaminase (ADA, EC 3.5.4.4), known as adenosine aminohydrolase, is a key enzyme engaged in purine metabolism that irreversibly converts adenosine to inosine or 2′deoxyadenosine to 2′deoxyinosine
[1]
. The amino acid sequence of ADA is highly conserved between species and it has a wide phylogenetic distribution from bacteria to humans
. In human tissues, adenosine deaminase is present as two isoenzymes: ADA1 and ADA2.
[4]
ADA1 constitutes the majority of ADA activity and it is present in virtually all tissues, with the highest levels in lymphoid cells, red blood cells and endothelium
. In addition to cytosolic form, ADA1 can be found as an ecto-enzyme binding to the cell surface by CD26 protein or adenosine receptors
[6]
. The second iso-enzyme (ADA2) belongs to the adenosine deaminase growth factor family and it is found with ADA1 mainly in monocytes/macrophages
[7]
. Zavialov et al. revealed that similarly to ADA1, ADA2 can be bind to the cell surface by proteoglycans or adenosine receptors
[8]
. As both isoenzymes of ADA maintain their catalytic activities after cleavage from the plasma membrane of intact cells submitted to stressful conditions, this makes them capable of deaminating adenosine into inosine in the soluble fraction of a given tissue, even if it is located away from the originating cell.
[9]
It has been shown that both isoenzymes, but with a predominance of ADA2, are present in human plasma in the soluble forms
[10]
. Plasma of rat and mouse are totally devoid of ADA2, where entire adenosine deaminating activity derives from ADA1
[7]
. The differentiation of ADA1 and ADA2 isoenzymes has been possible since ADA1 activity is inhibited by (+)-erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) and its derivatives, whereas ADA2 activity is not affected by these inhibitors
[11]
. The inhibition of ADA1 by EHNA-derived compounds provides evidence of the difference in binding sites of EHNA in two ADA isoenzymes: the hydrophobic site on ADA1 that is required for complex formation with the aliphatic chain of EHNA is probably absent in ADA2
[12]
. In humans, the ADA1-like domain of the ADA2 iso-enzyme shares about 70% of its amino acid similarity with the ADA1 protein
[13]
.
Intracellularly, adenosine deaminase plays a significant and diverse metabolic role. The importance of normal ADA activity levels is demonstrated by the effect of a genetic deficiency of this enzyme, which is associated with a form of severe combined immunodeficiency disease (SCID).
[14]
Due to the lack of ADA activity, 2′deoxyadenosine accumulates and is converted to 2′-deoxy-adenosine-5′-triphosphate (dATP), which inhibits ribonucleotide reductase (RNR), a crucial enzyme in DNA synthesis that follows disrupted T-cell development
[15]
. In physiology, concentration of adenosine, the second substrate for ADA, is maintained inside the cell on the low level by adenosine kinase with Km value ~ 1 µM
[16]
. However, during inflammation or hypoxia, adenosine is produced from intracellular ATP under conditions of low energy charge. AMP, which originates from ATP degradation is rapidly transformed to adenosine that is not immediately deaminated to inosine due to high Km of ADA (25–100 µM)
[17]
. This results in temporary accumulation of adenosine, which is vigorously exported out the cell via equilibrative nucleoside transporters (ENTs)
[18]
.
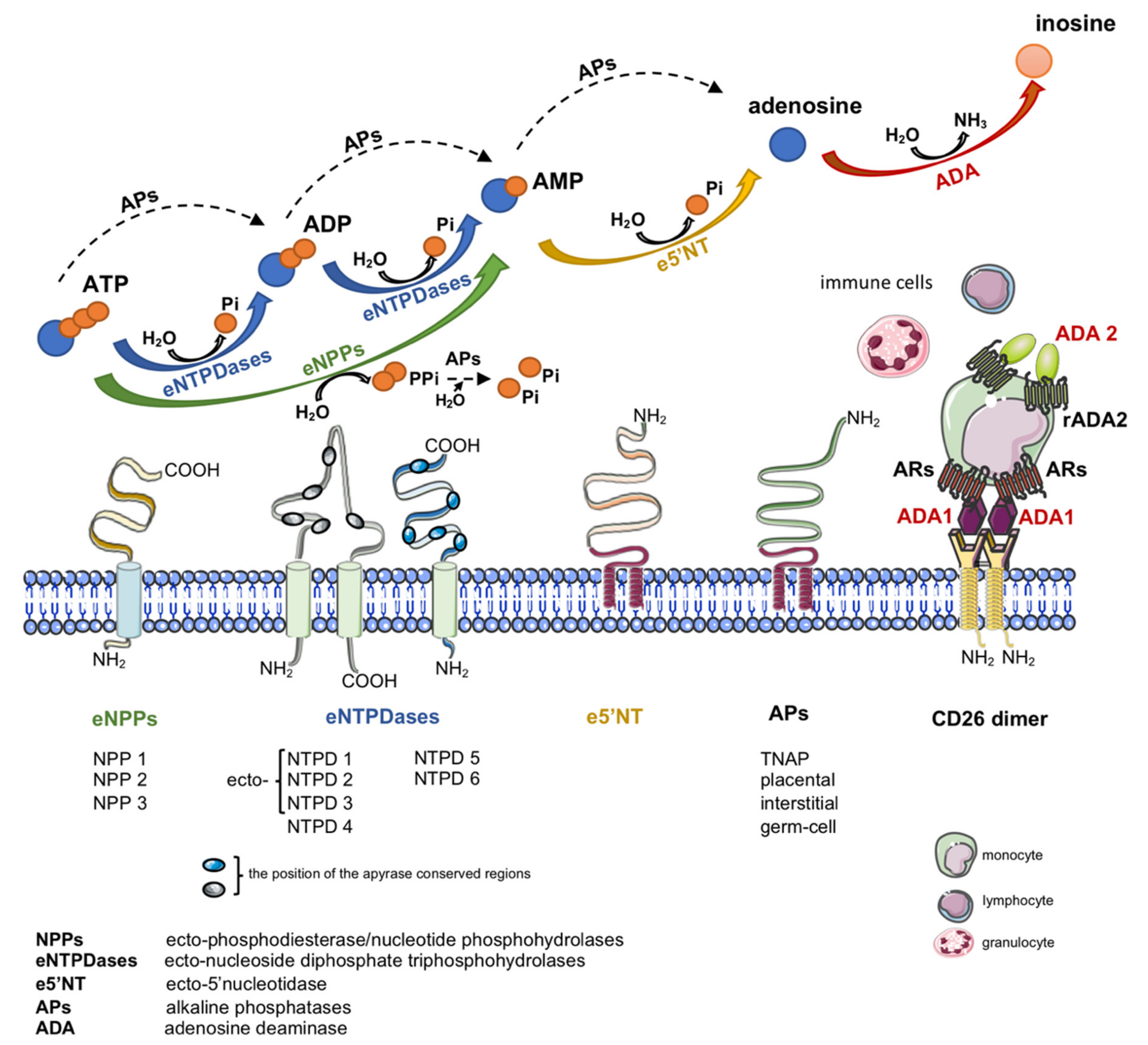
Another important source of extracellular adenosine is the hydrolysis of adenine nucleotides by cell-surface ecto-nucleotidases (Figure 1)
[19]
. Nucleotides are released to the extracellular compartment in response to cellular stress via lytic and non-lytic mechanisms
[20]
. Therefore, together with nucleotide-degrading ecto-enzymes and transmembrane adenosine transport, eADA plays an essential role in the regulation of extracellular adenosine concentration.
2. Adenosine
Adenosine is a ubiquitous molecule with key regulatory and cytoprotective mechanisms. Among a plethora of physiological actions, extracellular adenosine has an important role in cardiovascular system homeostasis
[21]
. It engages members of the G-protein coupled adenosine receptor (AR) family to mediate mostly beneficial adaptive and acute responses within all constituent cells of the heart and vasculature. In this way the four AR subtypes-A1, A2A, A2B, and A3Rs-regulate myocardial contraction, heart rate and conduction, adrenergic control, coronary vascular tone, cardiac and vascular growth, inflammatory-vascular cell interactions, and cellular stress-resistance, injury and death
[22]
. All subtypes of AR are characterized by seven transmembrane domains connected by three intracellular and extracellular domains. At the extracellular level, the N-terminus presents specific glycosylation sites, while at the intracellular side, the C-terminus contains phosphorylation and palmitoylation sites, important for receptor desensitization
[23]
. Each AR has unique cell and tissue distribution, secondary signaling transductors, affinity for adenosine, and physiological effects. In cardiovascular system, all AR subtypes were demonstrated in endothelium, vascular smooth muscle cells, cardiomyocytes, valve cells and many others
. A1R and A3R predominantly activate heterotrimeric G proteins belonging to the Gα i/o family, which inhibit cAMP production by adenylate cyclase, in contrast A2AR and A2BR predominantly activate Gαs family members, which stimulate cAMP production
[25]
.
The A1, A2A and A3 receptors possess high affinity for adenosine while the A2B shows relatively lower affinity for this nucleoside. During normal conditions, the extracellular adenosine accumulation is limited to 30–300 nM range activating only high-affinity AR
[26]
. Mounting evidence indicates that extracellular adenosine levels increase dramatically to micromolar concentrations that activate low-affinity receptors in tissues submitted to stressful conditions, such as ischemia, hypoxia, and inflammation, being a key protective and anti-inflammatory mechanism
[27]
. In addition to adenosine, it has been shown that other molecules can non-selectively activate adenosine receptors with micromolar affinities, including natural plant alkaloids such as caffeine, theophylline and their derivatives
[28]
. Particularly interesting is the fact that also inosine can interact with AR, which emphasizes the importance of eADA as controlling the ratio between extracellular adenosine and inosine
[29]
. From one side, inosine at micromolar concentrations expressed immunomodulatory and anti-inflammatory effects via A1R, A2AR and A3R
. While from the other, the opposed effects of inosine and adenosine have been demonstrated. In the study of Herman-de-Sousa et al.
[32]
adenosine favored normal collagen production by human subcutaneous fibroblasts via A2AR, whereas, inosine via A3R stimulated inappropriate dermal remodeling, providing ADA inhibition as a therapeutic target to prevent connective tissue disorganization. It is worth noting that inosine at micromolar concentrations stimulates the receptors with high affinity for adenosine, which may be a compensatory anti-inflammatory mechanism in the case of increased eADA activity. In addition, increased eADA expression and hence the amplified association of eADA with AR can allosterically modulate agonist affinity and efficacy
[33]
.
Except its enzymatic function, cell-surface eADA plays a significant extra-enzymatic role in the interactions between cells that expressed ADA-anchoring proteins on their surfaces (Figure 1). Indeed, there is an evidence of the formation of trimeric complexes dipeptidyl peptidase IV (CD26)-ADA-A2AR involving two cells
[6]
. This co-stimulatory and cell-to-cell connecting actions of eADA along with its activity regulate many cellular processes related to proliferation and differentiation, which affect pathological conditions associated with cardiovascular diseases such as endothelial activation and dysfunction, inflammation, myocardial ischemia-reperfusion injury or coagulation disorders. Since these pathologies are associated with ADA overexpression, the inhibition of its activity as well as binding to the surface proteins exhibit an attractive therapeutic potential in cardiovascular diseases. There are several groups of ADA inhibitors and some of them are presently used in clinical practice, however they still show side effects. Therefore, new ADA inhibitors that are devoid of cytotoxic effects are currently intensively studied. Based on above considerations, the present article has been conceived to provide a systematic review of compounds inhibiting ADA activity and its interactions with cell surface proteins as well as to summarize current information on the cardiovascular pathologies with ADA overactivity.
3. Conclusions
Adenosine deaminase is a moonlighting protein that exhibits intracellular and extracellular enzymatic activity, which modulates adenosine and 2′-deoxyadenosine concentration inside and outside the cell. Moreover, it has extra-enzymatic, co-stimulatory properties. Therefore, adenosine deaminase is an essential regulator of multiple cellular processes, including DNA synthesis, adenosine receptor-mediated pathways and cell-to-cell interactions. Overactivities of total-, soluble- and ecto- adenosine deaminase have been described in many cardiovascular pathologies (Table 1) including atherosclerosis, thrombosis, hypertension, myocardial ischemia-reperfusion injury, and diabetes, stimulating their further development.
Therefore, the inhibition of adenosine deaminase expression and activity (particularly its cell-surface and soluble forms) has been proposed as a promising cardioprotective therapy. Although the most popular adenosine deaminase inhibitors, such as 2′deoxycoformycin and EHNA exhibited beneficial effects for the development and progression of cardiovascular diseases, they have problems with poor pharmacokinetics and several toxicities related to their permeability through the cell membrane. So far, 2′deoxycoformycin is the only adenosine deaminase inhibitor in clinical use that currently is limited to the treatment of adult patients with hairy cell leukemia. Unfortunately, its acid-lability results in the lack of oral bioavailability and imposes its intravenous administration. Therefore, the development of novel cell non-permeable adenosine deaminase inhibitors that would have adequate pharmacokinetic properties is urgently needed.
Table 1. Cardiovascular diseases with ADA overactivity and therapeutic effects of its inhibition in experimental models. AMI—acute myocardial injury; IRI—ischemia-reperfusion injury; T2DM—type 2 diabetes mellitus; tADA—total adenosine deaminase; ADA1—adenosine deaminase 1; ADA2—adenosine deaminase 2; dCF—2′deoxycoformycin; EHNA—erythro-9-(2-hydroxy-3-nonyl) adenine; n.d.—no data.
| Cardiovascular Pathology | ADA Activity | ADA Inhibitor | Therapeutic Effect of ADA Inhibition |
|---|---|---|---|
|
Cardiovascular Pathology |
ADA Activity |
ADA Inhibitor |
Therapeutic Effect of ADA Inhibition |
| Atherosclerosis | dCF [35] | + | |
|
Atherosclerosis | |||
| ↑ tADA (plasma) | [34] ↑ ADA1 (vessel wall) [35] | ||
|
↑ tADA (plasma) [34]↑ ADA1 (vessel wall) [35] |
dCF [35] |
+ |
|
| Thrombosis | ↑ tADA (plasma) [36] | n.d. | n.d. |
|
Thrombosis |
↑ tADA (plasma) [36] |
n.d. |
n.d. |
| AMI/IRI | ↑ tADA (plasma) [37] | ||
|
AMI/IRI |
↑ tADA (plasma) | ||
| [ | 38 | ||
| 37 | |||
| ] | dCF [39] | + | |
| [ | 38] |
dCF [39] |
+ |
| Hypertension | ↑ tADA (plasma) [34] | EHNA [40] | + |
|
Hypertension |
↑ tADA (plasma) [34] |
EHNA [40] |
+ |
| T2DM | ↑ tADA (plasma) [41] ↑ ADA1 (plasma) [42] | ||
|
T2DM |
↑ tADA (plasma) [41]↑ ADA1 (plasma) | ||
| [ | 43 | ||
| 42 | |||
| ] | ↑ ADA2 (plasma) | ||
| [ | 43] | ||
| [ | 42] | ||
↑ ADA2 (plasma) | |||
| [ | 43 | ||
| 42 | |||
| ] | dCF [44] | + | |
| [ | 43] |
dCF [44] |
References
- Cortés, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluís, C.; Canela, E.I.; Casadó, V. Moonlighting Adenosine Deaminase: A Target Protein for Drug Development. Med. Res. Rev. 2015, 35, 85–125.
- Chang, Z.Y.; Nygaard, P.; Chinault, A.C.; Kellems, R.E. Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residues: Implications for catalytic function. Biochemistry 1991, 30, 2273–2280.
- Holm, L.; Sander, C. An evolutionary treasure: Unification of a broad set of amidohydrolases related to urease. Proteins 1997, 28, 72–82.
- Kutryb-Zajac, B.; Mierzejewska, P.; Sucajtys-Szulc, E.; Bulinska, A.; Zabielska, M.A.; Jablonska, P.; Serocki, M.; Koszalka, P.; Milczarek, R.; Jasztal, A.; et al. Inhibition of LPS-stimulated ecto-adenosine deaminase attenuates endothelial cell activation. J. Mol. Cell. Cardiol. 2019, 128.
- Ebrahimi-Rad, M.; Khatami, S.; Ansari, S.; Jalylfar, S.; Valadbeigi, S.; Saghiri, R. Adenosine Deaminase 1 as a Biomarker for Diagnosis and Monitoring of Patients with Acute Lymphoblastic Leukemia. J. Med. Biochem. 2018, 37, 128–133.
- Moreno, E.; Canet, J.; Gracia, E.; Lluís, C.; Mallol, J.; Canela, E.I.; Cortés, A.; Casadó, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A2A Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106.
- Kaljas, Y.; Liu, C.; Skaldin, M.; Wu, C.; Zhou, Q.; Lu, Y.; Aksentijevich, I.; Zavialov, A.V. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell. Mol. Life Sci. 2017, 74, 555–570.
- Zavialov, A.V.; Engström, A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem. J. 2005, 391, 51–57.
- Correia-de-Sá, P.; Adães, S.; Timóteo, M.A.; Vieira, C.; Magalhães-Cardoso, T.; Nascimento, C.; Duarte-Araújo, M. Fine-tuning modulation of myenteric motoneurons by endogenous adenosine: On the role of secreted adenosine deaminase. Auton. Neurosci. 2006, 126–127, 211–224.
- Gao, Z.-W.; Wang, X.; Lin, F.; Dong, K. Total adenosine deaminase highly correlated with adenosine deaminase 2 activity in serum. Ann. Rheum. Dis. 2020.
- Tardif, V.; Muir, R.; Cubas, R.; Chakhtoura, M.; Wilkinson, P.; Metcalf, T.; Herro, R.; Haddad, E.K. Adenosine deaminase-1 delineates human follicular helper T cell function and is altered with HIV. Nat. Commun. 2019, 10, 1–15.
- Andreasyan, N.A.; Hairapetyan, H.L.; Sargisova, Y.G.; Mardanyan, S.S. ADA2 isoform of adenosine deaminase from pleural fluid. FEBS Lett. 2005, 579, 643–647.
- Zavialov, A.V.; Yu, X.; Spillmann, D.; Lauvau, G.; Zavialov, A.V. Structural Basis for the Growth Factor Activity of Human Adenosine Deaminase ADA2. J. Biol. Chem. 2010, 285, 12367–12377.
- Bradford, K.L.; Moretti, F.A.; Carbonaro-Sarracino, D.A.; Gaspar, H.B.; Kohn, D.B. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID): Molecular Pathogenesis and Clinical Manifestations. J. Clin. Immunol. 2017, 37, 626–637.
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanetj. Rare Dis. 2018, 13, 1–7.
- Köse, M.; Schiedel, A.C.; Bauer, A.A.; Poschenrieder, H.; Burbiel, J.C.; Akkinepally, R.R.; Stachel, H.-D.; Müller, C.E. Focused screening to identify new adenosine kinase inhibitors. Bioorg. Med. Chem. 2016, 24, 5127–5133.
- Bin, A.; Caputi, V.; Bistoletti, M.; Montopoli, M.; Colucci, R.; Antonioli, L.; De Martin, S.; Castagliuolo, I.; Orso, G.; Giaroni, C.; et al. The ecto-enzymes CD73 and adenosine deaminase modulate 5′-AMP-derived adenosine in myofibroblasts of the rat small intestine. Purinergic Signal. 2018, 14, 409–421.
- Pastor-Anglada, M.; Pérez-Torras, S. Who Is Who in Adenosine Transport. Front. Pharmacol. 2018, 9, 627.
- Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784.
- Soslau, G. Extracellular adenine compounds within the cardiovascular system: Their source, metabolism and function. Med. Drug Discov. 2019, 4, 100018.
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243.
- Headrick, J.P.; Ashton, K.J.; Rose′meyer, R.B.; Peart, J.N. Cardiovascular adenosine receptors: Expression, actions and interactions. Pharmacol. Ther. 2013, 140, 92–111.
- Ballesteros-Yáñez, I.; Castillo, C.A.; Merighi, S.; Gessi, S. The role of adenosine receptors in psychostimulant addiction. Front. Pharmacol. 2018.
- Kutryb-Zajac, B.; Jablonska, P.; Serocki, M.; Bulinska, A.; Mierzejewska, P.; Friebe, D.; Alter, C.; Jasztal, A.; Lango, R.; Rogowski, J.; et al. Nucleotide ecto-enzyme metabolic pattern and spatial distribution in calcific aortic valve disease; its relation to pathological changes and clinical presentation. Clin. Res. Cardiol. 2020.
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264.
- Choudhury, H.; Chellappan, D.K.; Sengupta, P.; Pandey, M.; Gorain, B. Adenosine Receptors in Modulation of Central Nervous System Disorders. Curr. Pharm. Des. 2019, 25, 2808–2827.
- Grenz, A.; Homann, D.; Eltzschig, H.K. Extracellular adenosine: A safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxid Redox Signal 2011, 15, 2221–2234.
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 151–199.
- Welihinda, A.A.; Kaur, M.; Greene, K.; Zhai, Y.; Amento, E.P. The adenosine metabolite inosine is a functional agonist of the adenosine A2A receptor with a unique signaling bias. Cell. Signal. 2016, 28, 552–560.
- Da Rocha Lapa, F.; de Oliveira, A.P.L.; Accetturi, B.G.; de Oliveira Martins, I.; Domingos, H.V.; de Almeida Cabrini, D.; de Lima, W.T.; Santos, A.R.S. Anti-inflammatory effects of inosine in allergic lung inflammation in mice: Evidence for the participation of adenosine A2A and A 3 receptors. Purinergic Signal. 2013, 9, 325–336.
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489.
- Herman-de-Sousa, C.; Pinheiro, A.R.; Paramos-de-Carvalho, D.; Costa, M.A.; Ferreirinha, F.; Magalhães-Cardoso, T.; Ribeiro, S.; Pelletier, J.; Sévigny, J.; Correia-de-Sá, P. Opposing Effects of Adenosine and Inosine in Human Subcutaneous Fibroblasts May Be Regulated by Third Party ADA Cell Providers. Cells 2020, 9, 651.
- Gracia, E.; Farré, D.; Cortés, A.; Ferrer-Costa, C.; Orozco, M.; Mallol, J.; Lluís, C.; Canela, E.I.; McCormick, P.J.; Franco, R.; et al. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. Faseb J. 2013, 27, 1048–1061.
- Sajjan, N.B.; Makandar, A. Evaluation of serum adenosine deaminase levels with components of metabolic syndrome. Orig. Res. Artic. Int. J. Clin. Biochem. Res. 2016, 3, 285–291.
- Kutryb-Zajac, B.; Mateuszuk, L.; Zukowska, P.; Jasztal, A.; Zabielska, M.; Toczek, M.; Zakrzewska, A.; Sitek, B.; Rogowski, J.; Lango, R.; et al. Increased activity of vascular adenosine deaminase in atherosclerosis and therapeutic potential of its inhibition. Cardiovasc. Res. 2016, 112, 590–605.
- Stafford, N.P.; Pink, A.E.; White, A.E.; Glenn, J.R.; Heptinstall, S. Mechanisms Involved in Adenosine Triphosphate–Induced Platelet Aggregation in Whole Blood. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1928–1933.
- Patil, N.; Chavan, V.; Karnik, N.D. Antioxidant status in patients with acute myocardial infarction. Indian J. Clin. Biochem. 2007, 22, 45–51.
- Jordan, J.E.; Zhao, Z.Q.; Vinten-Johansen, J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc. Res. 1999, 43, 860–878.
- Oladipo, O.O.; Afolabi, B.B.; Okorodudu, A.O. Adenosine Deaminase Activity in Subjects With Normal Pregnancy, Pregnancy Induced Hypertension and Pre-Eclampsia. West Afr. J. Med. 2009, 28.
- Xia, C.; Rao, X.; Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J. Diabetes Res. 2017, 2017, 6494795.
- Sapkota, L.B.; Thapa, S.; Subedi, N. Correlation study of adenosine deaminase and its isoenzymes in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2017, 5, e000357.
- Larijani, B.; Heshmat, R.; Ebrahimi-Rad, M.; Khatami, S.; Valadbeigi, S.; Saghiri, R. Diagnostic Value of Adenosine Deaminase and Its Isoforms in Type II Diabetes Mellitus. Enzyme Res. 2016, 2016, 9526593.
- Takhshid, M.A.; Zahediannejad, Z.; Aboualizadeh, F.; Moezzi, L.; Ranjbaran, R. G22A Polymorphism of Adenosine Deaminase and its Association with Biochemical Characteristics of Gestational Diabetes Mellitus in an Iranian Population. Iran. J. Med. Sci. 2015, 40, 170–174.
- Zhong, J.; Gong, Q.; Goud, A.; Srinivasamaharaj, S.; Rajagopalan, S. Recent Advances in Dipeptidyl-Peptidase-4 Inhibition Therapy: Lessons from the Bench and Clinical Trials. J. Diabetes Res. 2015, 2015, 1–14.