1. Introduction
Non-Invasive Brain Stimulation (NIBS) techniques, including transcranial Direct Current Stimulation (tDCS), transcranial Alternating Current Stimulation (tACS) and repetitive Transcranial Magnetic Stimulation (rTMS), have been proposed for years to improve both motor and non-motor symptoms in a number of neurological conditions, comprising neurodegenerative disorders as Alzheimer’s (AD) and Parkinson’s Disease (PD)
[1][2][3][4][5][1,2,3,4,5]. They are safe and promising tools for the modulation of cortical and, probably, sub-cortical activities
[6]. A growing body of literature strengthens their use for the treatment of both speech disturbances and axial symptoms of PD (bradykinesia, falls and dysphagia), where conventional pharmacological approaches did not provide long-lasting changes over time
[7]. Moreover, they proved a significant effect for the treatment of the so-called “freezing of gait” (FOG), which still remains a challenge for clinicians and neuroscientists
[8][9][10][11][8,9,10,11]. However, there is a substantial lack of papers discussing their putative role as disease-modifying, neuroprotective therapies; this is of key importance because pharmacological treatments show merely a “symptomatic” effect, without any significant interference with disease progression over time
[12]. In this re
svie
arch, the researchers ew, we encompass current knowledge about NIBS and neuroprotection, discussing novel data and old concepts, both in animal and human models, and highlighting the possible use of these techniques in early phases of the neurodegenerative process. Moreover,
theywe suggest a new view of tDCS and rTMS mechanisms of action, not based on either polarity or frequency-dependence of their after-effects, but on their ability to interfere with pathological protein accumulation and degradation in experimental models of neurodegenerative disorders. As a matter of debate and to give a more complete picture about neurostimulation and neuroprotection,
theywe briefly provide a comparison between NIBS and invasive brain stimulation, as DBS (“Deep Brain Stimulation”), towards the goal of neuroprotection, both in degenerative and non-degenerative disorders; accordingly, DBS has recently demonstrated interesting results in animal models of schizophrenia and depression
[13][14][13,14].
2. NIBS and Neuroprotection in Parkinson’s Disease
2.1. tDCS
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting about 1% of the population >60 years of age
[15]. The pathological hallmark of PD is the progressive degeneration of dopaminergic neurons in the substantia nigra and striatum
[16], ultimately leading to motor (e.g., tremor, akinesia, rigidity, gait impairments) and non-motor (e.g., anxiety, depression, cognitive deficits) symptoms. However, the underpinning mechanisms remain unclear
[17]. Multiple factors may contribute to neuronal damage, both in mutation related and in idiopathic forms of the disease
[18], including biochemical factors, causing cellular stress accumulation, due to inflammation, oxidative stress and excitotoxicity, and leading to mitochondrial dysfunction, energy production loss and cell demise (e.g., mitochondrial dysfunction, defective protein degradation, neuroinflammation, oxidative stress, excitotoxicity), all of which are tightly linked to each other
[19][20][21][19,20,21]. Available treatments are only symptomatic, and pharmacological therapies lead to several and disabling side effects over time
[12].
Several neuromodulation techniques have been suggested, such as complementary treatment approach, both invasive
[22][23][24][22,23,24] and non-invasive
[25][26][25,26]. Among these, transcranial direct current stimulation (tDCS) showed a convincing therapeutic potential, with benefits both on motor and cognitive performances
[27][28][29][30][31][32][33][27,28,29,30,31,32,33]. However, the mechanisms by which tDCS exerts its effects in PD patients are not fully understood, particularly at cellular and molecular levels
[34]. Current knowledge suggests that tDCS increases the release of dopamine
[35][36][37][35,36,37], modulates alpha-synuclein aggregation and autophagic degradation
[34], alters neurotransmitters concentration (e.g., GABA, serotonin, glutamate)
[38] and induces anti-apoptotic and anti-inflammatory effects
[20][39][20,39]. However, these cellular effects have been collected either in vitro or in animal models, but not yet confirmed in human studies. In this scenario, tDCS is likely to enhance the expression of brain-derived neurotrophic factor (BDNF)
[40], a neurotransmitter modulator and neurotrophic factor that supports neurogenesis
[41][42][41,42] and survival of neurons
[43]. Therefore, these findings suggest a possible neuroprotective effect of tDCS, which appears to be partially effective in restoring some of the biochemical defects associated with neurodegenerative diseases, as confirmed by animal studies
[17][19][44][45][17,19,44,45] (see
Figure 1). Indeed, current knowledge comes from neurotoxin-treated animal models, and shows preliminary and promising results in terms of tDCS-induced antioxidant function and survival of dopaminergic cells from neurotoxin-induced cell death
[17][19][44][45][17,19,44,45].
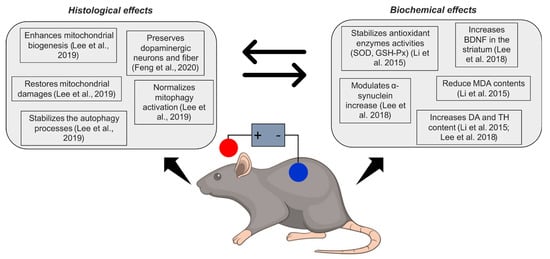
Figure 1. Schematic overview of neuroprotective effects of tDCS in animal models of Parkinson’s disease. SOD = superoxide dismutase; GSH-Px = glutathione peroxidase; BDNF = brain-derived neurotrophic factor; MDA = malonaldehyde; DA = dopamine; TH = tyrosine hydroxylase. Feng et al., 2020
[44]; Lee et al., 2019
[17]; Li et al., 2015
[19]; Lee et al., 2018
[45].
In PD patients, the alteration of tyrosine hydroxylase activity (TH—an enzyme catalysing the precursor of dopamine, L-DOPA, in dopamine) reduces dopamine (DA) levels
[46]. Besides, oxidative stress is increased and antioxidative processes are inhibited
[19]. tDCS may have a role in neuronal protection acting on the response against oxidative stress, as suggested by Lee et al., 2019
[17] and Li et al., 2015
[19] (see
Table 1). Anodal tDCS applied on mice preserves dopaminergic neurons after the injection of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
[17] and increases both DA and TH content
[19]. Also, it reduces the decrease of antioxidant enzymes activities (superoxide dismutase, SOD; and glutathione peroxidase, GSH-Px) induced by MPTP, ultimately improving the survival response in the nigral-striatal area. However, antioxidative results might not be a direct effect of such a response and may not be driven directly by the stimulation, but rather a consequence of enhanced secretion of BDNF induced by tDCS, as shown by previous studies
[38].
Studies assessing neuroprotective effects of tDCS in animal models of Parkinson’s disease.
The major target of oxidative stress is the mitochondrion-increased production (or decreased capacity to eliminate) of free radicals, known to induce neuronal death via disruption of mitochondrial function
[47]. Several findings suggest a pivotal role of mitochondrial dysfunctions in PD pathogenesis
[48]. Lee et al., 2019
[17] demonstrated in mice that anodal tDCS exerts a neuroprotective effect against MPTP toxicity, by normalizing mitophagy activation, enhancing mitochondrial biogenesis and restoring mitochondrial damage (see
Table 1)
[17][49][17,49]. tDCS also decreases the effects of MPTP, i.e., increased expression of mitophagy-related proteins (PTEN-induced putative kinase 1, PINK1; Parkin; and microtubule-associated protein light chain 3, LC3), PINK1/Parkin upregulation and enhanced autophagic flux
[17]. Mitochondrial biogenesis-related proteins (peroxisome proliferator-activated receptor γ coactivator, PGC1α; and nuclear respirator factor 1, NRF1) and mitochondrial transcription factor A (TFAM) were increased by tDCS, suggesting that its biogenetic effect might be exerted at the level of transcription and replication of mitochondrial DNA
[17]. Also, tDCS recovered an MTPT-induced increase of dynamin-related protein 1 (Drp1) expression, which reflects mitochondrial fragmentation and the release of pro-apoptotic proteins
[50].
In PD, oxidative stress (OxS), mitochondrial dysfunction, excitotoxicity, and neuroinflammation are strictly linked to autophagy pathways. Autophagy is a cellular homeostatic process involved in both unspecific bulk degradation (macroautophagy, referred to simply as “autophagy”) of cytosolic proteins, aggregates and organelles
[51] but also in specific catabolism (chaperone-mediated autophagy, CMA) of neuropathological proteins, including alpha-synuclein
[52]. Defects in macroautophagy and CMA have been shown to play an important role in the pathogenesis of the disease
[53]. To date, while no data are available in the literature on the specific effect of electrical stimulation on CMA, with the only exception of a recent study from
the our
esearchers' group
[34], studies have investigated the effect on macroautophagy. Lee et al., 2018
[45] demonstrated that anodal tDCS over the left motor cortical area stabilizes the autophagy processes activated by MPTP-induced toxicity, as showed by microtubule-associated protein 1 light chain 3 (LC3) and AMP-activated protein kinase (AMPK) upregulation, and the mechanistic target of rapamycin (mTOR) and sequestosome1/p62 (p62) downregulation in experimental mice (see
Table 1). However, such an effect has not been described for MTPT treatment-free, suggesting that the effect of anodal tDCS might occur under stress conditions. Also, anodal tDCS modulates the MPTP-induced upregulation of α-synuclein in substantia nigra pars compacta, which has been identified as a distinctive marker for PD
[54]. As for the antioxidative effects, however, it is still under debate whether these effects on autophagy are a direct effect of tDCS, or rather a result of an increased release of BDNF
[55].
Overall, these results represent a theoretical basis for the study of tDCS (anodal polarity) as a potential neuroprotective rather than a symptomatic therapy, as has mostly been considered so far. This application would be of great interest, since there is an absolute unmet need for treatments aiming to halt or restore the disease.
2.2. rTMS
A recent study evidenced a neuroprotective effect of early rTMS, as suggested by its ability to preserve tyrosine hydroxylase- (TH-) positive neurons in the substantia nigra pars compacta (SNpc) and fibers in the striatum in a hemiparkinsonian rat model induced by unilateral injection of 6-hydroxydopamine (6-OHDA)
[56]. Furthermore, a previous study performed in parkinsonian rats induced by the inhibition of the ubiquitin-proteasome system, another catabolic pathway different from autophagy, whose dysfunction also exerts a pathogenic role in PD, demonstrated that rTMS exerts neuroprotective effects by alleviating the loss of TH-positive dopaminergic neurons, by preventing the loss of striatal dopamine levels, by reducing the levels of apoptotic protein (cleaved caspase-3) and inflammatory factors (cyclooxygenase-2 and tumor necrosis factor alpha) in lesioned substantia nigra
[57].
3. NIBS and Neuroprotection in Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder clinically characterized by amnestic and non-amnestic cognitive impairments. In pathological neurons, β-amyloid (Aβ)-containing extracellular plaques and tau-containing intracellular neurofibrillary tangles determine the aggregation of misfolded proteins, which leads to microtubule disorganization, cholinergic dysfunction, neuroinflammation, OxS, and, ultimately, neural dysfunction and synaptic loss
[58]. Current pharmacological treatments for AD are mostly symptomatic, and therapies altering the underlying pathological processes are not commonly available. Similar to PD, non-invasive brain stimulation (NIBS) techniques (e.g., tDCS, transcranial magnetic stimulation—TMS) were shown to improve AD symptoms (e.g., global cognition, cognitive and memory functions, executive performance)
[59][60][59,60] (see
Figure 2). Several randomized clinical trials have been conducted by using either tDCS or rTMS for the treatment of cognitive symptoms associated to AD
[61][62][63][61,62,63]. However, to date, the biochemical mechanisms are still not fully understood (see
Table 2). Neurotrophic factors (NTFs) regulate the growth, survival, proliferation, migration and differentiation of neurons
[64] and have been extensively studied in the context of AD. In AD, the lowered expression of NTFs, such as nerve growth factor (NGF)
[65], BDNF
[66], glial cell line-derived neurotrophic factor (GDNF)
[67] and ciliary neurotrophic factor (CNTF), have been observed in affected brain regions, including the temporal cortex and hippocampus
[68]. Recently, particular interest has been aroused by the potentially beneficial effect of neuromodulation techniques on BDNF, which is required in the hippocampus for late-phase long-term potentiation and represents one of the most important cellular mechanisms that underlies learning and memory (see
Table 2). Moreover, BDNF induces the secretion of acetylcholine by enhancing the differentiation and survival of cholinergic neurons in the basal forebrain
[69]. Notably, various studies have recently shown increasing BDNF levels in the basal forebrain and hippocampus in AD animal models
[70] as well as in the serum of AD patients
[71] after rTMS when compared to controls. Similarly, rTMS was found to be effective on NGF brain levels
[72][73][72,73]. Moreover, rTMS and tDCS were effective also on the BDNF-TrkB signalling pathway
[38][55][74][38,55,74], which affects cell survival, migration, outgrowth of axons and dendrites, synaptogenesis, synaptic transmission, and synapse remodelling
[75].
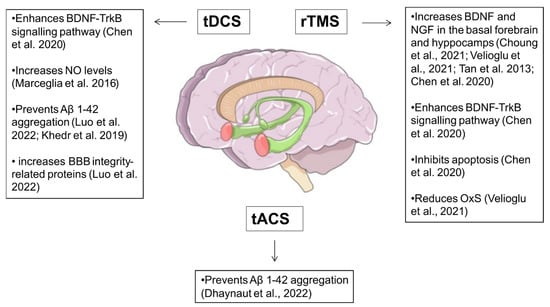
Figure 2. Schematic overview of neuroprotective effects of NIBS in Alzheimer’s disease. tDCS = transcranial direct current stimulation; tACS = transcranial alternating current stimulation; rTMS = repetitive transcranial magnetic stimulation; BDNF = brain-derived neurotrophic factor; TrkB = Tropomyosin receptor kinase B; NGF = Nerve growth factor; NO = nitric oxide; OxS = oxidative stress. Choung et al., 2021
[70]; Velioglu et al., 2021
[71]; Tan et al., 2013
[73]; Chen et al., 2020
[74]; Marceglia et al., 2016
[76][83]; Luo et al., 2022
[77][88]; Khedr et al., 2019
[78][90].
Studies assessing neuroprotective effects of NIBS in animal models of Alzheimer’s Disease (AD).
Beyond enhancing neuron survival, rTMS and tDCS concurrently inhibit apoptosis
[79][80][76,77]. Specifically, TMS effectively balanced the apoptotic pathways in an AD-mice model
[72], by inhibiting pro-apoptotic members of the Bcl-2 family, such as cleaved caspase-3 and Bax, which are usually overexpressed in AD. Conversely, the same
res
earchtudy pointed out TMS-induced reduced ubiquitination of beta catenin, which notoriously promotes cell survival.
Brain tissue in AD patients is characterized by increased oxidative stress (OxS), due to an imbalance in Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) levels and the antioxidant defense system, resulting in damage to proteins, lipids, and DNA oxidation/glycoxidation processes
[81][78]. Velioglu and co-workers pointed out beneficial effects of rTMS on oxidative stress levels in AD patients by applying rTMS over the lateral parietal cortex
[71]. In AD, OxS contributes to endothelial Nitric Oxide (NO) depletion
[82][83][79,80] and quickening cognitive decline
[84][81] due to cerebral hypoperfusion. Conversely, cerebral hypoperfusion is responsible for increased OxS. Trivedi et al.
[85][82] applied low electrical fields to endothelial cells to induce the increase of NO levels, and, in turn, vasodilatation. Similarly, Marceglia et al.
[76][83] speculated that tDCS may raise the NO level to prevent brain and hippocampal hypoperfusion. Further studies investigated NIBS’s beneficial effect on brain perfusion via blood-brain barrier modulation. Neurons, glial cells and cerebral blood vessels are closely related in structure and function, collectively referred to as the “neurovascular unit” (NVU), which is critical for regulating cerebral blood flow, maintaining both the blood-brain barrier integrity and signal transduction between cells. In AD brains, there is evidence that the cerebral microcirculation system is damaged and the main components of NVU underwent pathological changes
[86][84]. tDCS has been demonstrated to decrease the number of glial cells and increase levels of vascular endothelial growth factor (VEGF) and interleukin-8, possibly resulting in decreased local inflammation, increased vascularization, improved toxic metabolite clearance and microcirculation protection
[87][88][89][85,86,87]. A further study
[77][88] showed that tDCS-treated AD model mice exhibited reduced glial fibrillary acidic protein (GFAP) levels. GFAP plays a crucial role in endothelial junction function and morphologic changes of astrocyte end foot processes
[90][89]. NIBS might also prevent Aβ 1-42 aggregation, increasing Aβ serum levels and this was assessed by both tDCS in AD patients
[78][90] and tACS in AD-mouse model
[91], presumably via microglia activation
[92].