Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alla Mitrofanova and Version 2 by Sirius Huang.
Diabetes is the leading cause of chronic kidney disease worldwide. Despite the burden, the factors contributing to the development and progression of diabetic kidney disease (DKD) remain to be fully elucidated. Increasing evidence suggests that mitochondrial dysfunction is a pathological mediator in DKD as the kidney is a highly metabolic organ rich in mitochondria. Furthermore, low-grade chronic inflammation also contributes to the progression of DKD, and several inflammatory biomarkers have been reported as prognostic markers to risk-stratify patients for disease progression and all-cause mortality.
- DKD
- inflammation
- mitochondria
- innate immunity
- podocyte
- tubule cells
1. cGAS-STING Signaling
Cyclic GMP-AMP (cGAS) is a nuclear and cytosolic protein that senses the presence of double-stranded DNA in the cytosol leading to the formation of a second messenger, cyclic GMP-AMP (cGAMP) and activation of stimulator of interferon genes (STING). STING activation culminates in the recruitment of different kinases: TANK-binding kinase 1 (TBK1), mitogen-activated protein kinase kinase kinase 14 (MAP3K14 or NIK) and heterotrimeric IκB kinase (IKK), which, in turn, promote interferon regulatory factor 3 (IRF3), non-canonical nuclear factor kappa B (NF-κB) and canonical NF-κB, respectively. As a result, IRF3 activation results in type I interferon responses, which are usually mainly associated with antiviral and anticancer effects [1][18], while NF-κB activation may lead to a broad spectrum of effects [2][3][4][19,20,21]. A common concept suggests that STING predominantly localizes to the outer membrane of the endoplasmic reticulum, but some studies report the presence of STING in the mitochondrial membrane [5][6][22,23].
While the cGAS-STING pathway was originally discovered in the context of the innate immunity response to infections and cancer [7][8][9][24,25,26], it is clear that the STING pathway is more than just important in pathogen detection; it also plays a significant role in the detection of self-DNA released from damaged mitochondria (Figure 1), dying cells or tumor cells. Thus, activation of the cGAS-STING in response to the presence of mitochondrial DNA (mtDNA) present in the cytosol of cells has been shown in mouse models of renal fibrosis [10][27]. Studies suggest that STING phosphorylation is increased in the db/db mouse model of DKD at baseline and that pharmacological STING inhibition protects from DKD progression [11][12][28,29]. Interestingly, induction of STING itself in wildtype mice resulted in proteinuria and podocyte foot process effacement in studies [11][12][28,29]. Using eNOS db/db mice and rats with type 2 diabetic nephropathy, others demonstrated increased activity of the cGAS-STING pathway [13][30]. In patients with DKD, the presence of plasma and urinary mtDNA was recently recognized as a potential marker of early DKD progression [14][15][16][17][31,32,33,34]. However, the mechanisms of mtDNA escape into the cytosol remain elusive.
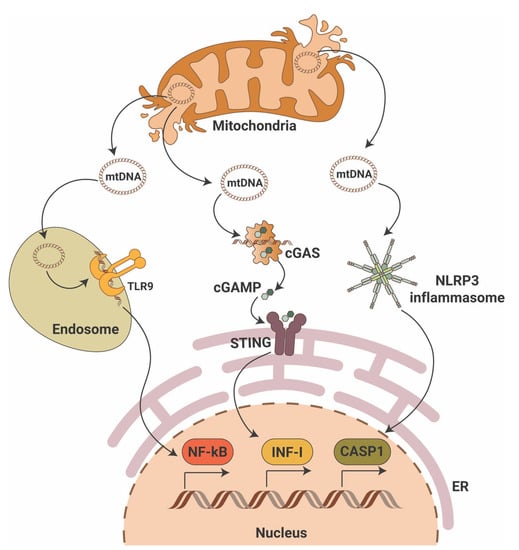
Figure 1. Mitochondrial DNA triggers pro-inflammatory signaling pathways. Mitochondrial DNA (mtDNA) can activate three major pro-inflammatory signaling pathways that include endosomal toll-like receptor 9 (TLR9) with activation of nuclear factor κB (NF-κB), the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon gene (STING) with activation of interferon type I (INF-I) and cytosolic NLR family pyrin domain containing 3 (NLRP3) inflammasome activation with induction of caspase-1-dependent apoptosis. ER—endoplasmic reticulum; cGAMP—2′3′-cyclic GMP-AMP.
mtDNA is particularly vulnerable to damage. Given the fact that mtDNA resides in close proximity to mitochondrial reactive oxygen species (mtROS), oxidative lesions often cause mtDNA damage via DNA strand breakage or nucleotide base oxidation. In DKD, chronic hyperglycemia disrupts the bioenergetic balance, which results in mtROS production and oxidative mtDNA damage as shown in studies of glomeruli from DBA/2J mice [18][35] and streptozotocin (STZ)-induced DKD rats [19][36]. Additionally, in STZ-induced DKD mice mitochondrial biogenesis is reduced due to decreased mRNA expression of peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α), nuclear respiratory factor 1 (NRF-1) and mitochondrial transcriptional factor A (TFAM) [20][37]. Interestingly, decreased expression of TFAM has been reported in renal tissues from patients with CKD stage 4, while TFAM knockdown in mice results in mtDNA leakage into the cytosol and activation of the cGAS-STING pathway [10][27]. Release of mtDNA into the cytosol can cause activation of the proapoptotic pore-forming proteins BCL2-associated X, apoptosis regulator (BAX) and BCL2 antagonist/killer 1(BAK1) as discussed below. A link between lipotoxicity and mtDNA release into the cytosol followed by activation of the cGAS-STING pathway was recently reported in db/db mice on a high fat diet [21][38].
In summary, an abundant literature shows the ability of mtDNA to activate the cGAS-STING signaling pathway in the kidney, thereby driving an inflammatory response in DKD.
2. RLR Signaling
The RIG-I like receptor (RLR) signaling pathway is another inflammatory pathway that can be stimulated by mitochondria (Figure 1). Unlike the cGAS-STING pathway, the RLR pathway is activated by foreign, altered, or ectopic RNA [22][39]. It has been shown that mitochondrial RNA (mtRNA) can be released into cytosol in a state of reduced polyribonucleotide nucleotidyltransferase 1 (PNPT1) expression, leading to mtRNA degradation, and activation of RLR melanoma differentiation-associated protein 5 (MDA5) [23][40]. Interestingly, mtDNA double-strand breaks have been shown to contribute to BAX-BAK1-mediated mtRNA release into cytosol with further activation of RIG-I, but not MDA5 [24][41]. However, the mechanisms leading to the differential activation of RIG-I versus MDA5 by mtRNA species remain to be discovered.
Notably, a recent study of patients with type 2 diabetes revealed a specific serum RNA signature in patients with DKD when compared to patients with diabetes and no DKD, where downregulation of four mitochondrial messenger RNAs (ATP6, ATP8, COX3 and ND1) was found to correlate with serum creatinine and estimated glomerular filtration rate [25][42]. A genome-wide association study analysis of patients with type 1 diabetes (n = 19,406) also revealed the association of RIG-I/MDA5 and interferon alpha beta gene set [26][43].
3. Inflammasome Signaling
The inflammasome comprises a class of signalosomes in innate immunity that promote inflammation and induce an inflammatory form of programmed cell death, called pyroptosis. Early studies have shown that cytosolic mtDNA can also drive the activation of inflammasomes [27][44], specifically of the inflammasome that contains nucleotide-binding domain-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) as a sensing component (Figure 1). NLRP3 assembles the inflammasome through oligomerization with apoptosis-associated speck-like protein (ASC) to elicit robust caspase 1 (CASP1) activation and production of interleukins 1β (IL-1β) and 18 (IL-18). Oxidized mtDNA release into the cytosol upon mitochondrial dysfunction has been shown to activate the NLRP3 inflammasome [28][45], and a feedforward loop was identified in which inflammasome activation facilitates mtDNA release via mtROS production [29][46]. Besides the mtROS-associated NLRP3-ASC-driven mtDNA escape into the cytosol, a physical interaction between NLRP3 and thioredoxin-interacting protein (TXNIP), a nuclear protein that controls the cellular redox state, may induce mitochondrial damage and mtDNA leakage [30][47]. However, ROS inhibitors seem to disrupt inflammasome priming, i.e., the synthesis of inflammasome components, but not its activation [31][48]. In line with this notion, recent studies showed that NLRP3 inflammasome activation depends on the mitochondrial electron transport chain [32][49], and that cytosolic oxidized mtDNA serves as the ultimate NLRP3 ligand [33][50]. Of note, mitochondrial damage per se does not trigger NLRP3 signaling if priming is omitted (reviewed in [34][51]). Moreover, NLRP3 inflammasome activation fails under conditions of TFAM deficiency [33][50]. However, mtDNA has been shown to also activate inflammasomes that use the absence of melanoma 2 (AIM2) as a sensing component [35][52]. Interestingly, while oxidized DNA seems to activate NLRP3 inflammasomes [28][45], AIM2-containing counterparts are suggested to recognize non-oxidized DNA [33][50].
Persistent and aberrant NLRP3 signaling underlies many chronic diseases, including type 1 and type 2 diabetes, while NLRP3 deficiency has been shown to protect against injury, irrespective of the renal cell type [36][37][53,54]. mtDNA seems to be one of the main triggers of NLRP3 activation in streptozotocin-induced diabetic [38][39][55,56] and in high fat diet mice [40][57]. Using primary renal tubular epithelial cells and unilateral ureteral obstruction (UUO) mice, a recent study demonstrated that peroxisomal proliferator-γ coactivator-1α (PGC-1α) ameliorates NLRP3 inflammasome-associated renal fibrosis via the modulation of mitochondrial dynamics [41][58]. NLRP3 activation also contributes to DKD progression as shown in a study demonstrating that podocyte-specific Nlrp3 or caspase-1 deficiency resulted in protection from DKD [42][59]. In contrast, another group reported that using an NLRP3-specific inhibitor, MCC950, did not confer renoprotective effects using streptozotocin-induced diabetic mice as it did not reduce renal inflammation (glomerular accumulation of CD68 positive cells), mesangial expansion and glomerulosclerosis [43][60]. However, an earlier study reported that MCC950 lowered fibrosis, renal inflammation and provided protection from kidney failure in a model of oxalate nephropathy [44][61]. Less is known about the role of AIM2 inflammasomes in diabetes and DKD development. It has been shown that AIM2 inflammasomes directly interact with apoptosis-associated speck-like protein and contribute to the development of many human diseases, including type 2 diabetes, where cell-free mtDNA has been shown to activate AIM2 inflammasomes [45][62].
In summary, mtDNA is a major DAMP for inflammasome activation that contributes to chronic kidney disease development and progression. Moreover, NLRP3 and AIM2 may represent a potential therapeutic target to ameliorate DKD-associated podocyte and tubular injury.
4. TLR Signaling
Toll-like receptor (TLR) signaling plays a key role in the innate immune system by recognizing pathogen-associated molecular patterns (PAMPs) leading to the activation of NF-κB and interferon production. The TLR family comprises 10 members in humans (TLR1-TLR10) and 12 members in mice (TLR1-TLR12). TLRs are located on the cell plasma membrane, except for TLR3, TLR7, TLR8 and TRL9, which are found in intracellular vesicles where they sense the nucleic acids inside a cell. Early studies showed that naked as well as protein-bound mtDNA has been shown to activate TLR9 (Figure 1) and advanced glycosylation end product-specific receptor (RAGE) [46][47][63,64]. Other studies demonstrated that treatment in vitro or in vivo with mtDNA results in increased levels of TLR9, NF-κB and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκB-α) in different tissues [48][49][50][65,66,67]. De novo TLR9 expression has been shown in podocytes of some patients with glomerular diseases [51][52][53][54][68,69,70,71], suggesting that endogenous mtDNA serves as a ligand and may facilitate podocyte apoptosis [55][72]. In a model of acute kidney injury, absence of TLR9 reduced mtDNA-mediated kidney injury [56][73]. In DKD, the expression of TLR2, TLR4, TLR5, TLR7, TLR8 and TLR9 has been described, but TLR2 and TLR4 are the two most extensively studied receptors (reviewed in [57][74]). Sparse studies on TLR3 and TLR9 in DKD suggest that in the ApoE-/- streptozotocin-induced mouse model of DKD, TLR3 and TLR9 are activated in the kidney [58][75]. Similarly, enhanced expression of TLR3 was reported in tubules from patients with DKD [59][76]. Nevertheless, no data are available that would connect activation of TLR3 and TLR9 in DKD with the release of mtDNA into the cytosol, which may be the subject of future studies.
5. NF-κB Signaling
Nuclear factor-κB (NF-κB) represents a family of transcription factors which consists of five structurally related members (NF-κB1, NF-κB2, RelA, RelB and c-Rel) and regulates a large array of genes involved in the regulation of the immune and inflammatory responses. The activation of the NF-κB involves two major signaling pathways: (1) in the canonical pathway, NF-κB responds to diverse stimuli, including cytokine receptors, PRRs, TNF receptors, T-cell and B-cell receptors [60][77]; (2) in the noncanonical (alternative) pathway, NF-κB selectively responds to specific ligands such as lymphotoxin beta receptor (LTβR), tumor necrosis factor receptor superfamily member 13C (TNFRSF13C or BAFFR), CD40, or RANK [61][78]. Functionally, the canonical NF-κB pathway is involved in almost all aspects of the immune response, while the noncanonical NF-κB appears to be involved in the regulation of specific functions of the adaptive immune system. Interestingly, the suppression of inhibitor of apoptosis (IAP) proteins by the cytosolic mitochondrial protein SMAC shifts NF-κB signaling from the canonical to the noncanonical pathway upon stabilization of mitogen-activated protein kinase kinase kinase 14 (MAP3K14), and this process is orchestrated by BAK1-BAX oligomers [62][79].
A recent study addressing the role of NF-κB in DKD showed an activation of the NF-κB pathway in diabetic rats that progress to DKD, whereby inflammation was restricted to the glomerular compartment with intense glomerular macrophage infiltration [63][80]. Earlier studies also confirmed modest activation of the glomerular NF-κB signaling pathway in streptozotocin treated rats as early as one month after the induction of diabetes [64][81], while in patients with type 2 diabetes, NF-κB activation was mainly detected in cortical tubular epithelial cells and, to a lesser extent, in some glomeruli [65][82]. Similarly, in patients with type 1 diabetes and DKD, p65 positive glomeruli and inflammation in the area of the renal interstitium were found [63][80]. High glucose has also been shown to induce NF-κB activation and upregulation of proinflammatory cytokines in human proximal tubular epithelial cells [66][83] and in podocytes [67][84]. Interestingly, long-term (12 months) NF-κB inhibition in diabetic rats using pyrrolidine dithiocarbamate resulted in reduced IL-6 production and prevented the development of glomerulosclerosis and loss of podocyte integrity in one study [63][80]. Therefore, the canonical pathway seems to be the prevalent NF-κB activation pathway in DKD (Figure 2). However, the exact mechanisms leading to NF-κB activation in DKD remain unclear and require further investigation.
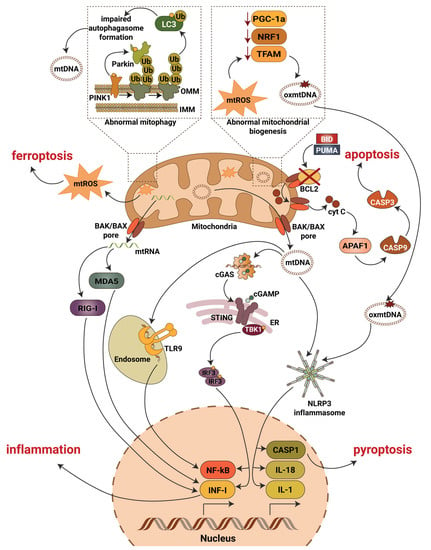
Figure 2. Mitochondrial contribution to DKD associated kidney damage. Generation of mitochondrial reactive oxygen species (mtROS), escape of mitochondrial RNA (mtRNA) and mitochondrial DNA (mtDNA) into cytosol, production of oxidized mitochondrial DNA (oxmtDNA) and abnormalities in mitophagy lead to activation of major pro-inflammatory signaling pathways, pyroptotic, apoptotic and ferroptotic cell death. Abbreviations: APAF1—apoptotic pepdidase activating factor 1; BAK—Bcl2 homologous antagonist/killer; BAX—Bcl2-associated X protein; BCL2—B-cell lymphoma 2; BID—BH3 interacting-domain death agonist; CASP1—caspase 1; CASP3—caspase 3; CASP9—caspase 9; cGAS—cyclic GMP-AMP synthase; cGAMP—2′3′-cyclic GMP-AMP; Cyt C—cytochrome c; ER—endoplasmic reticulum; IL-1—interleukin 1; IL-18—interleukin 18; IMM—inner mitochondrial membrane; INF-I—interferon type I; IRF3—interferon regulatory factor 3; LC3—microtubule-associated protein 1A/1B-light chain 3; MDA5—melanoma differentiation-associated protein 5; NF-kB—activation of nuclear factor κB; NLRP3—NLR family pyrin domain containing 3; NRF1—nuclear respiratory factor 1; OMM—outer mitochondrial membrane; PGC-1a—peroxisome proliferator-activated receptor-γ coactivator-1α1; PINK1—PTEN induced kinase 1; PUMA—p53 upregulated modulator of apoptosis; RIG-I—retinoic acid-inducible gene I; STING—stimulator of interferon gene; TBK1—TANK binding kinase 1; TFAM—mitochondrial transcription factor A; TLR9—toll-like receptor 9; Ub—ubiquitin.
To make the picture even more complicated, NF-κB subunits (IkBα, p65) and NF-κB pathway proteins (IKKα, IKKβ and IKKγ) are present in the inner mitochondria matrix [68][69][70][71][85,86,87,88]. Collectively, these studies suggest that NF-κB can non-specifically bind mtDNA sequences and regulate mRNA expression of a variety of target genes. Recent studies also demonstrated that NF-κB is involved in mitochondrial fission [72][89], regulation of BAX mediated cytochrome c release to control apoptosis [73][90], organization of the energy metabolism network by controlling the balance between glycolytic utilization and mitochondrial respiration [25][74][42,91], and in controlling respiratory chain gene expression including the expression of COXI, COXIII and CytB [69][75][76][86,92,93]. Moreover, NF-κB p62 induction restricts NLRP3 inflammasome activation via the elimination of damaged mitochondria [77][94].