Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by 潘 丛彬.
Actin participates in the formation of highly differentiated myofibrils under the regulation of actin-binding proteins (ABPs), which provides a structural basis for the contractile function and morphological change in cardiomyocytes.
- actin-binding proteins
- cardiac hypertrophy
- F-actin
- fetal genes
1. Introduction
The microfilament cytoskeleton is mainly composed of actin and actin-binding proteins (ABPs). Actin is one of the most abundant cytoskeletal proteins in eukaryotes and is involved in cell morphology change, migration, division and other cellular processes [9,10][1][2]. Actin takes two forms in cells: actin monomers (also known as globular actin, G-actin) and actin filaments (also known as filamentous actin, F-actin). Actin dynamics are finely regulated by a variety of ABPs (Table 1) [11][3]. Actin is involved in the formation of sarcomeres in cardiomyocytes [12][4]. The straight and uniform sarcomeric F-actin is critical for the contractile function of muscle [13][5]. In addition, actin assembly is thought to be related with autophagy [14,15][6][7]. The inhibition of F-actin disassembly can suppress autophagosome formation [16][8]. Several studies have found that F-actin is significantly accumulated abnormally in hypertrophic cardiomyocytes [17,18,19][9][10][11]. The dysregulation of F-actin accumulation may lead to cardiac hypertrophy through disrupting autophagy and sarcomeric structure. The function of ABPs in the development of cardiac hypertrophy has been gradually elucidated.
Table 1.
Actin-binding proteins.
| Types | ABPs | Basic Function | Refs. |
|---|---|---|---|
| G-actin-binding | Profilin, thymosin β4, cofilin | Bound to G-actin | [11,20,21][3][12][13] |
| F-actin-binding | Dystrophin, tropomyosin | Bound to F-actin | [9,11,22][1][3][14] |
| Actin-nucleating | Formin, Arp2/3 complex, proteins with tandem WH2 domains, leiomodin | Nucleation to initiate actin polymerization | [11,23,3][15]24,[16]25][[17] |
| Actin-elongating | Formin, tetramers of Ena/VASP | Regulation of actin assembly | [11,24][3][16] |
| Actin-bundling | Fimbrin/Plastin, hhLIM, gelsolin | Causes parallel F-actin filaments to closely pack together | [26,27,28,29][18][19][20][21] |
| Severing | ADF/cofilin, gelsolin, twinfilin, FRL-α, INF-2 | Severs F-actin | [30,31,][22][32,23][33,24][25]34[26] |
| Capping | Twinfilin, gelsolin, tropomodulin, CapZ, Arp2/3 complex | Caps F-actin to inhibit actin polymerization | [11,35,36,37][3][27][28][29] |
| Motor | Myosin | Cargo transfer | [38][30] |
2. ABPs in Cardiac Hypertrophy
2.1. Profilin-1
Profilin is widely expressed in most eukaryotes and has a molecular weight of about 17 kDa [39][31]. There are various profilin isoforms expressed in different tissues. Profilin-1 is universally expressed, profilin-2 is specifically expressed in the brain and profilin-3 and profilin-4 are specifically expressed in kidney and testis, respectively [40][32]. Profilin accelerates the nucleotide exchange of G-actin and delivers ATP-G-actin to the growing barbed ends of F-actin through interacting with the poly-proline motifs of formin, vasodilator-stimulated phosphoprotein (VASP) and CDC42-activated Wiskott Aldrich syndrome protein (WASP)/WASP family [20,41,42,43][12][33][34][35]. Profilin-1 is directly associated with cardiac hypertrophy [44][36]. Overexpression of profilin-1 in the vascular tissues of FVB/N mice leads to vascular remodeling and hypertension by increasing actin aggregation, which provides mechanical stress for the development of cardiac hypertrophy [45,46][37][38]. It has been shown that the protein level of profilin-1 is significantly increased in mammalian hypertrophic hearts (Figure 21). The myocardin-related transcription factor megakaryoblastic leukemia (MKL) induces the expression of the signal transducer and activator of transcription 1 (STAT1) via its SAP-domain (SAF-A/B, acinus and PIAS protein domain) activity, which upregulates PFN expression [47][39]. Whether this is the explanation for the increased protein level of profilin in cardiac hypertrophy remains to be investigated. In cardiomyocytes, the functional abnormality of profilin-1 can change the abundance or activity of multiple proteins associated with cardiomyopathy. For example, the overexpression of profilin-1 can contribute to decreases in the phosphorylation level of endothelial nitric oxide synthases (eNOS) at Ser1177 in the hearts of spontaneous hypertensive rats [17][9]. Levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), skeletal muscle α-actin (α-SMA) and phosphorylated ERK1/2 (active form) were significantly increased in neonatal rat ventricular myocytes (NRVMs) following stimulation by phenylephrine or endothelin 1, which can be inhibited by siRNA-directed PFN1 silencing [44][36]. Increased phosphorylation of ERK1/2 activates the mechanistic (mammalian) target of rapamycin complex 1 (mTORC1) that subsequently inhibits autophagy [48,49,50][40][41][42]. It may be a potential key mechanism of cardiac hypertrophy mediated by the dysregulation of profilin-1 (Figure 21). Additionally, the inhibition of Rho-associated coiled-coil-containing protein kinase pathway (ROCK) can suppress the upregulation of profilin-1 induced by advanced glycation end products (AEGs) in H9c2 cells [51][43]. By comparison, overexpression of PFN1 results in the reactivation of fetal genes (NPPA and NPPB), an increase in F-actin in myocardium and destruction of myofibrils [44][36]. These processes can be reversed by inhibiting the expression of profilin-1 [17][9]. The inhibition of profilin-1 expression in H9c2 cells and Sprague–Dawley rats can attenuate cardiac hypertrophy induced by AEGs [51,52][43][44]. In Drosophila, myocyte-specific overexpression of profilin leads to disorders in muscle fibers and sarcomeres, which result in damaged muscle ultrastructure and function [44][36].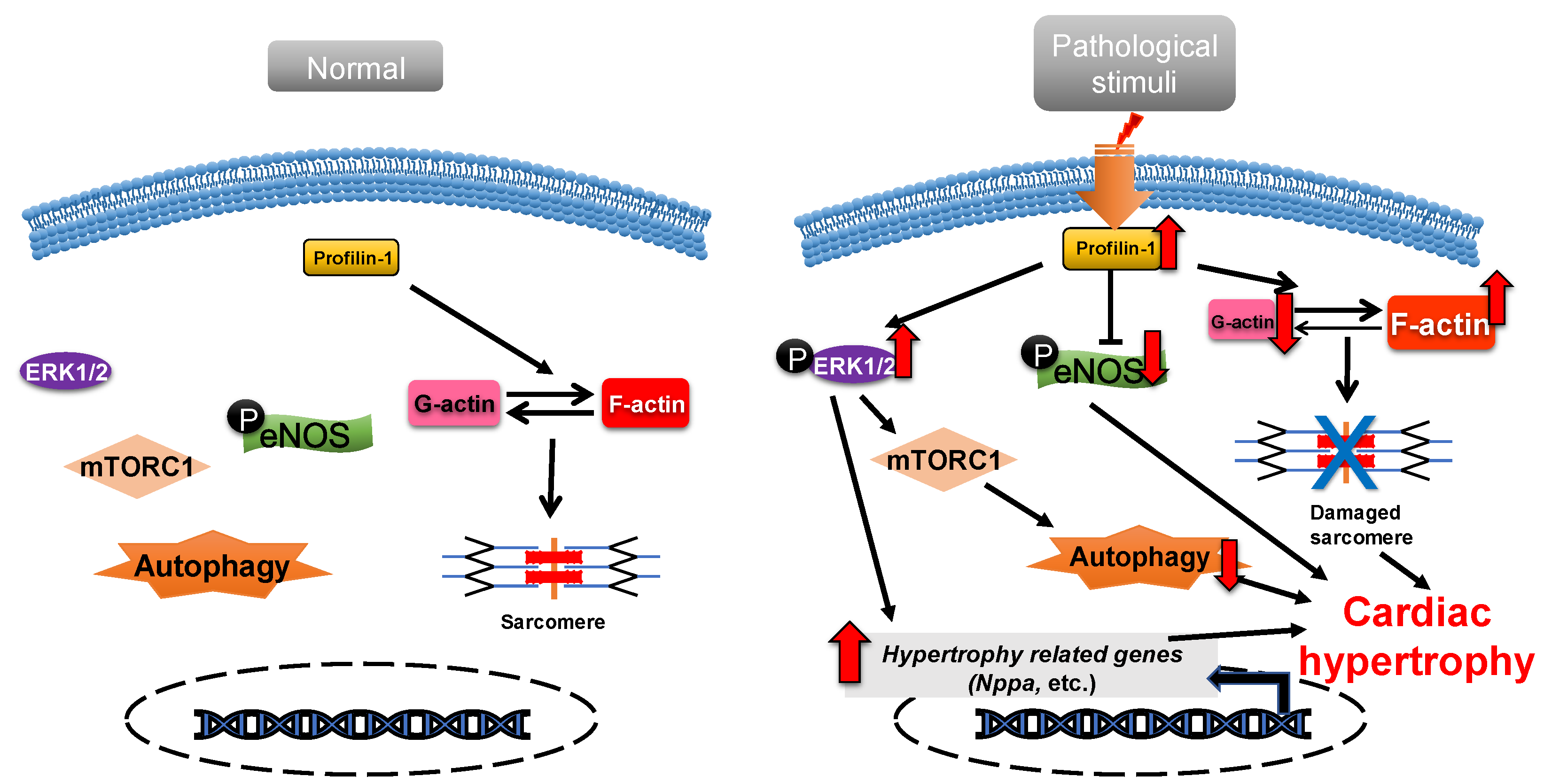
Figure 21. Profilin-1 mediates cardiac hypertrophy. In normal cardiomyocytes, profilin-1 is at a basal level and the fetal genes are not activated. Pathological stimuli increase the protein level of profilin-1, which results in ERK1/2 activation, F-actin accumulation and eNOS inhibition. This results in the reactivation of hypertrophy-related genes, inhibition of autophagy and damage to sarcomere structure and, ultimately, the development of cardiac hypertrophy.
2.2. ADF/Cofilin
Actin-depolymerizing factor (ADF)/cofilin consists of a single ADF homologous domain and has a molecular weight of about 15 kDa. The ADF/cofilin family contains ADF (also known as destrin, mainly expressed in endothelial and epithelial cells) and two cofilin isoforms (cofilin-1 is universal and cofilin-2 is cardio-specific) [53,54][45][46]. ADF/cofilin can bind to both G-actin and F-actin and can sever and depolymerize F-actin in regulating actin dynamics, which contributes to the cell contractility power [55][47]. The activity of cofilin is regulated by phosphorylation primarily from the ROCK/Lin-11, Isl1 and MEC-3 domain kinase (LIMK)/cofilin signaling pathway (Figure 32) [56,57][48][49]. Cofilin is inactivated via phosphorylation.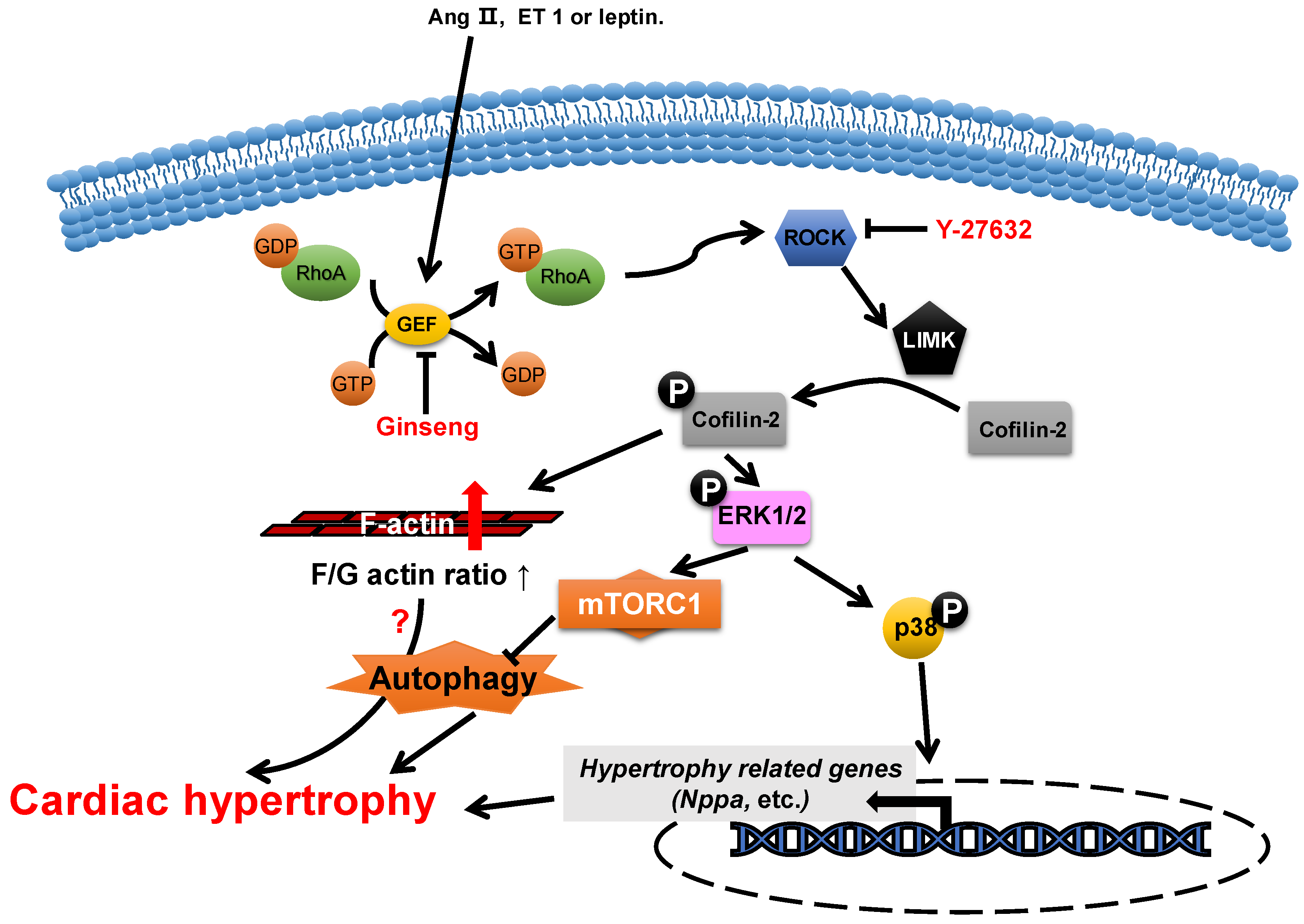
Figure 32. Proposed roles of cofilin-2 in cardiac hypertrophy. Neurohumoral factors (e.g., Ang II, ET 1 and leptin) lead to cofilin-2 phosphorylation through the RhoA/ROCK/LIMK signaling pathway. Phosphorylated cofilin-2 can lead to F-actin accumulation, which may subsequently contribute to cardiac hypertrophy through disrupting autophagy. In addition, it promotes the activation of ERK1/2 and p38, which contributes to the inhibition of autophagy and the reactivation of hypertrophy-related genes, which subsequently cause cardiac hypertrophy.
2.3. Formin
Formin is a type of multidomain protein consisting of 7 subfamilies and 15 members in human genes. Formins are characterized by the presence of two conserved domains: formin homology 1 (FH1) and FH2. FH1 binds to the profilin–actin complex via poly-proline sequences and brings the G-actin to FH2, which promotes actin nucleation and polymerization [11,65][3][57].2.4. CapZ
CapZ, a type of capping protein, anchors F-actin to the Z disc and regulates actin turnover, which contributes to sarcomere structural changes [76,77][58][59]. PIP2, a downstream effector of RAC1, can promote the dissociation of CapZ from F-actin by weakening their binding affinity [78,79,80][60][61][62]. Overexpression of CapZ in transgenic mice can lead to fatal cardiac hypertrophy [77][59]. It has been shown that hypertrophic agonists, phenylephrine or endothelin can reduce the binding affinity between CapZ and F-actin via PIP2-dependent pathways in NRVMs [81][63]. This may result in sarcomere remodeling, which induces cardiac hypertrophy. The cyclic mechanical strain activates downstream focal adhesion kinase (FAK) via the mechanotransduction of integrin, which then activates phosphatidylinositol 4-phosphate 5-kinase (PIP5K) through the RhoA/ROCK pathway. PIP5K phosphorylates phosphatidylinositol 4-phosphate (PI4P) in order to produce PIP2, which reduces the affinity of CapZ and F-actin binding, which contributes to the dysregulation of F-actin assembly and cardiac hypertrophy (Figure 53) [80,82,83,84][62][64][65][66].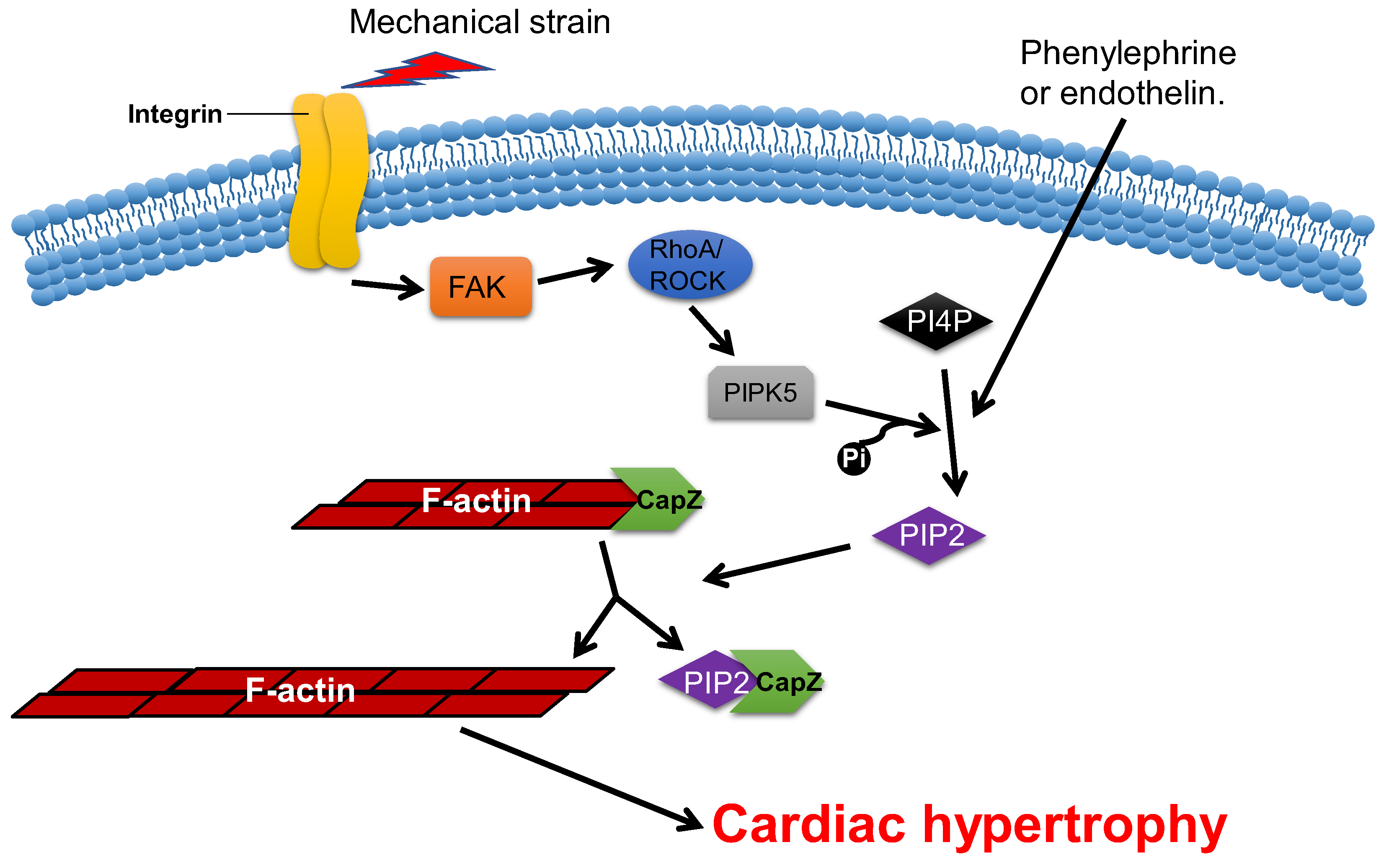
Figure 53. CapZ regulates cardiac hypertrophy. Mechanotransduction leads to the activation of RhoA/Rho-kinase pathway through integrins, which reduce the binding affinity of CapZ and F-actin. It subsequently causes cardiac hypertrophy.
References
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654.
- Chalut, K.J.; Paluch, E.K. The Actin Cortex: A Bridge between Cell Shape and Function. Dev. Cell 2016, 38, 571–573.
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226.
- Pluess, M.; Ehler, E. Cardiac Cytoarchitecture in Health and Disease. In Cardiac Cytoarchitecture: How to Maintain a Working Heart; Ehler, E., Ed.; Springer: Cham, Switzerland, 2015; pp. 1–14.
- Huang, X.; Li, Z.; Hu, J.; Yang, Z.; Liu, Z.; Zhang, T.; Zhang, C.; Yuan, B. Knockout of Wdr1 results in cardiac hypertrophy and impaired cardiac function in adult mouse heart. Gene 2019, 697, 40–47.
- Kruppa, A.J.; Kendrick-Jones, J.; Buss, F. Myosins, Actin and Autophagy. Traffic 2016, 17, 878–890.
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy 2012, 8, 1590–1603.
- Zhuo, C.; Ji, Y.; Chen, Z.; Kitazato, K.; Xiang, Y.; Zhong, M.; Wang, Q.; Pei, Y.; Ju, H.; Wang, Y. Proteomics analysis of autophagy-deficient Atg7-/- MEFs reveals a close relationship between F-actin and autophagy. Biochem. Biophys. Res. Commun. 2013, 437, 482–488.
- Zhao, S.H.; Qiu, J.; Wang, Y.; Ji, X.; Liu, X.J.; You, B.A.; Sheng, Y.P.; Li, X.; Gao, H.Q. Profilin-1 promotes the development of hypertension-induced cardiac hypertrophy. J. Hypertens. 2013, 31, 576–586; discussion 586.
- Zeidan, A.; Javadov, S.; Karmazyn, M. Essential role of Rho/ROCK-dependent processes and actin dynamics in mediating leptin-induced hypertrophy in rat neonatal ventricular myocytes. Cardiovasc. Res. 2006, 72, 101–111.
- Hunter, J.C.; Zeidan, A.; Javadov, S.; Kilic, A.; Rajapurohitam, V.; Karmazyn, M. Nitric oxide inhibits endothelin-1-induced neonatal cardiomyocyte hypertrophy via a RhoA-ROCK-dependent pathway. J. Mol. Cell. Cardiol. 2009, 47, 810–818.
- Funk, J.; Merino, F.; Venkova, L.; Heydenreich, L.; Kierfeld, J.; Vargas, P.; Raunser, S.; Piel, M.; Bieling, P. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife 2019, 8, e50963.
- Safer, D.; Elzinga, M.; Nachmias, V.T. Thymosin beta 4 and Fx, an actin-sequestering peptide, are indistinguishable. J. Biol. Chem. 1991, 266, 4029–4032.
- Rybakova, I.N.; Amann, K.J.; Ervasti, J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996, 135, 661–672.
- Cao, L.; Yonis, A.; Vaghela, M.; Barriga, E.H.; Chugh, P.; Smith, M.B.; Maufront, J.; Lavoie, G.; Méant, A.; Ferber, E.; et al. SPIN90 associates with mDia1 and the Arp2/3 complex to regulate cortical actin organization. Nat. Cell Biol. 2020, 22, 803–814.
- Kuhn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513.
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243.
- Hosokawa, N.; Kuragano, M.; Yoshino, A.; Shibata, K.; Uyeda, T.Q.P.; Tokuraku, K. Unidirectional cooperative binding of fimbrin actin-binding domain 2 to actin filament. Biochem. Biophys. Res. Commun. 2021, 552, 59–65.
- Sobral, A.F.; Chan, F.Y.; Norman, M.J.; Osorio, D.S.; Dias, A.B.; Ferreira, V.; Barbosa, D.J.; Cheerambathur, D.; Gassmann, R.; Belmonte, J.M.; et al. Plastin and spectrin cooperate to stabilize the actomyosin cortex during cytokinesis. Curr. Biol. 2021, 31, 5415–5428.e10.
- Zheng, B.; Wen, J.K.; Han, M. hhLIM is a novel F-actin binding protein involved in actin cytoskeleton remodeling. FEBS J. 2008, 275, 1568–1578.
- Archer, S.K.; Claudianos, C.; Campbell, H.D. Evolution of the gelsolin family of actin-binding proteins as novel transcriptional coactivators. BioEssays 2005, 27, 388–396.
- Chen, Q.; Courtemanche, N.; Pollard, T.D. Aip1 promotes actin filament severing by cofilin and regulates constriction of the cytokinetic contractile ring. J. Biol. Chem. 2015, 290, 2289–2300.
- Burtnick, L.D.; Urosev, D.; Irobi, E.; Narayan, K.; Robinson, R.C. Structure of the N-terminal half of gelsolin bound to actin: Roles in severing, apoptosis and FAF. EMBO J. 2004, 23, 2713–2722.
- Takács-Kollár, V.; Lőrinczy, D.; Nyitrai, M.; Hild, G. Spectroscopic characterization of the effect of mouse twinfilin-1 on actin filaments at different pH values. J. Photochem. Photobiol. B 2016, 164, 276–282.
- Harris, E.S.; Li, F.; Higgs, H.N. The mouse formin, FRLalpha, slows actin filament barbed end elongation, competes with capping protein, accelerates polymerization from monomers, and severs filaments. J. Biol. Chem. 2004, 279, 20076–20087.
- Gurel, P.S.; Ge, P.; Grintsevich, E.E.; Shu, R.; Blanchoin, L.; Zhou, Z.H.; Reisler, E.; Higgs, H.N. INF2-mediated severing through actin filament encirclement and disruption. Curr. Biol. 2014, 24, 156–164.
- Johnston, A.B.; Hilton, D.M.; McConnell, P.; Johnson, B.; Harris, M.T.; Simone, A.; Amarasinghe, G.K.; Cooper, J.A.; Goode, B.L. A novel mode of capping protein-regulation by twinfilin. eLife 2018, 7, e41313.
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 1999, 274, 33179–33182.
- Bao, Y.; Kake, T.; Hanashima, A.; Nomiya, Y.; Kubokawa, K.; Kimura, S. Actin capping proteins, CapZ (β-actinin) and tropomodulin in amphioxus striated muscle. Gene 2012, 510, 78–86.
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267.
- Pinto-Costa, R.; Sousa, M.M. Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton 2020, 77, 76–83.
- Jockusch, B.M.; Murk, K.; Rothkegel, M. The profile of profilins. Rev. Physiol. Biochem. Pharmacol. 2007, 159, 131–149.
- Suetsugu, S.; Miki, H.; Takenawa, T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998, 17, 6516–6526.
- Paul, A.S.; Pollard, T.D. The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 2008, 18, 9–19.
- Ferron, F.; Rebowski, G.; Lee, S.H.; Dominguez, R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007, 26, 4597–4606.
- Kooij, V.; Viswanathan, M.C.; Lee, D.I.; Rainer, P.P.; Schmidt, W.; Kronert, W.A.; Harding, S.E.; Kass, D.A.; Bernstein, S.I.; van Eyk, J.E.; et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 110, 238–248.
- Moustafa-Bayoumi, M.; Alhaj, M.A.; El-Sayed, O.; Wisel, S.; Chotani, M.A.; Abouelnaga, Z.A.; Hassona, M.D.; Rigatto, K.; Morris, M.; Nuovo, G.; et al. Vascular hypertrophy and hypertension caused by transgenic overexpression of profilin 1. J. Biol. Chem. 2007, 282, 37632–37639.
- Elnakish, M.T.; Hassanain, H.H.; Janssen, P.M. Vascular remodeling-associated hypertension leads to left ventricular hypertrophy and contractile dysfunction in profilin-1 transgenic mice. J. Cardiovasc. Pharmacol. 2012, 60, 544–552.
- Joy, M.; Gau, D.; Castellucci, N.; Prywes, R.; Roy, P. The myocardin-related transcription factor MKL co-regulates the cellular levels of two profilin isoforms. J. Biol. Chem. 2017, 292, 11777–11791.
- Oeing, C.U.; Nakamura, T.; Pan, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Kokkonen-Simon, K.M.; Lin, B.L.; Chen, A.; Zhu, G.; Bedja, D.; et al. PKG1alpha Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 2020, 127, 522–533.
- Altamirano, F.; Oyarce, C.; Silva, P.; Toyos, M.; Wilson, C.; Lavandero, S.; Uhlen, P.; Estrada, M. Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J. Endocrinol. 2009, 202, 299–307.
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269.
- Yang, D.; Wang, Y.; Jiang, M.; Deng, X.; Pei, Z.; Li, F.; Xia, K.; Zhu, L.; Yang, T.; Chen, M. Downregulation of Profilin-1 Expression Attenuates Cardiomyocytes Hypertrophy and Apoptosis Induced by Advanced Glycation End Products in H9c2 Cells. BioMed Res. Int. 2017, 2017, 9716087.
- Yang, D.; Liu, W.; Ma, L.; Wang, Y.; Ma, J.; Jiang, M.; Deng, X.; Huang, F.; Yang, T.; Chen, M. Profilin-1 contributes to cardiac injury induced by advanced glycation end-products in rats. Mol. Med. Rep. 2017, 16, 6634–6641.
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195.
- Nakashima, K.; Sato, N.; Nakagaki, T.; Abe, H.; Ono, S.; Obinata, T. Two mouse cofilin isoforms, muscle-type (MCF) and non-muscle type (NMCF), interact with F-actin with different efficiencies. J. Biochem. 2005, 138, 519–526.
- Mseka, T.; Cramer, L.P. Actin depolymerization-based force retracts the cell rear in polarizing and migrating cells. Curr. Biol. 2011, 21, 2085–2091.
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269.
- Hild, G.; Kalmár, L.; Kardos, R.; Nyitrai, M.; Bugyi, B. The other side of the coin: Functional and structural versatility of ADF/cofilins. Eur. J. Cell Biol. 2014, 93, 238–251.
- Rangrez, A.Y.; Hoppe, P.; Kuhn, C.; Zille, E.; Frank, J.; Frey, N.; Frank, D. MicroRNA miR-301a is a novel cardiac regulator of Cofilin-2. PLoS ONE 2017, 12, e0183901.
- Aoki, H.; Izumo, S.; Sadoshima, J. Angiotensin II activates RhoA in cardiac myocytes: A critical role of RhoA in angiotensin II-induced premyofibril formation. Circ. Res. 1998, 82, 666–676.
- Aikawa, R.; Komuro, I.; Nagai, R.; Yazaki, Y. Rho plays an important role in angiotensin II-induced hypertrophic responses in cardiac myocytes. Mol. Cell. Biochem. 2000, 212, 177–182.
- Zeidan, A.; Javadov, S.; Chakrabarti, S.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy involves selective caveolae and RhoA/ROCK-dependent p38 MAPK translocation to nuclei. Cardiovasc. Res. 2008, 77, 64–72.
- Moey, M.; Rajapurohitam, V.; Zeidan, A.; Karmazyn, M. Ginseng (Panax quinquefolius) attenuates leptin-induced cardiac hypertrophy through inhibition of p115Rho guanine nucleotide exchange factor-RhoA/Rho-associated, coiled-coil containing protein kinase-dependent mitogen-activated protein kinase pathway activation. J. Pharmacol. Exp. Ther. 2011, 339, 746–756.
- Zeidan, A.; Gan, X.T.; Thomas, A.; Karmazyn, M. Prevention of RhoA activation and cofilin-mediated actin polymerization mediates the antihypertrophic effect of adenosine receptor agonists in angiotensin II- and endothelin-1-treated cardiomyocytes. Mol. Cell. Biochem. 2014, 385, 239–248.
- Lai, D.; Gao, J.; Bi, X.; He, H.; Shi, X.; Weng, S.; Chen, Y.; Yang, Y.; Ye, Y.; Fu, G. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J. Mol. Med. 2017, 95, 155–165.
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74.
- Schafer, D.A.; Hug, C.; Cooper, J.A. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 1995, 128, 61–70.
- Hart, M.C.; Cooper, J.A. Vertebrate isoforms of actin capping protein beta have distinct functions In vivo. J. Cell Biol. 1999, 147, 1287–1298.
- Kim, K.; McCully, M.E.; Bhattacharya, N.; Butler, B.; Sept, D.; Cooper, J.A. Structure/function analysis of the interaction of phosphatidylinositol 4,5-bisphosphate with actin-capping protein: Implications for how capping protein binds the actin filament. J. Biol. Chem. 2007, 282, 5871–5879.
- Montgomery, D.E.; Chandra, M.; Huang, Q.; Jin, J.; Solaro, R.J. Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am. J. Physiol. Heart. Circ. Physiol. 2001, 280, H1011–H1018.
- Lin, Y.H.; Warren, C.M.; Li, J.; McKinsey, T.A.; Russell, B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell. Signal. 2016, 28, 1015–1024.
- Hartman, T.J.; Martin, J.L.; Solaro, R.J.; Samarel, A.M.; Russell, B. CapZ dynamics are altered by endothelin-1 and phenylephrine via PIP2- and PKC-dependent mechanisms. Am. J. Physiol. Cell Physiol. 2009, 296, C1034–C1039.
- Li, J.; Russell, B. Phosphatidylinositol 4,5-bisphosphate regulates CapZβ1 and actin dynamics in response to mechanical strain. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1614–H1623.
- Lin, Y.H.; Li, J.; Swanson, E.R.; Russell, B. CapZ and actin capping dynamics increase in myocytes after a bout of exercise and abates in hours after stimulation ends. J. Appl. Physiol. 2013, 114, 1603–1609.
- Li, J.; Mkrtschjan, M.A.; Lin, Y.H.; Russell, B. Variation in stiffness regulates cardiac myocyte hypertrophy via signaling pathways. Can. J. Physiol. Pharmacol. 2016, 94, 1178–1186.
More