Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by 潘 丛彬 and Version 2 by Camila Xu.
Actin participates in the formation of highly differentiated myofibrils under the regulation of actin-binding proteins (ABPs), which provides a structural basis for the contractile function and morphological change in cardiomyocytes.
- actin-binding proteins
- cardiac hypertrophy
- F-actin
- fetal genes
1. Introduction
The microfilament cytoskeleton is mainly composed of actin and actin-binding proteins (ABPs). Actin is one of the most abundant cytoskeletal proteins in eukaryotes and is involved in cell morphology change, migration, division and other cellular processes [1][2][9,10]. Actin takes two forms in cells: actin monomers (also known as globular actin, G-actin) and actin filaments (also known as filamentous actin, F-actin). Actin dynamics are finely regulated by a variety of ABPs (Table 1) [3][11]. Actin is involved in the formation of sarcomeres in cardiomyocytes [4][12]. The straight and uniform sarcomeric F-actin is critical for the contractile function of muscle [5][13]. In addition, actin assembly is thought to be related with autophagy [6][7][14,15]. The inhibition of F-actin disassembly can suppress autophagosome formation [8][16]. Several studies have found that F-actin is significantly accumulated abnormally in hypertrophic cardiomyocytes [9][10][11][17,18,19]. The dysregulation of F-actin accumulation may lead to cardiac hypertrophy through disrupting autophagy and sarcomeric structure. The function of ABPs in the development of cardiac hypertrophy has been gradually elucidated.
Table 1.
Actin-binding proteins.
| Types | ABPs | Basic Function | Refs. |
|---|---|---|---|
| G-actin-binding | Profilin, thymosin β4, cofilin | Bound to G-actin | [3][12][13][11,20,21] |
| F-actin-binding | Dystrophin, tropomyosin | Bound to F-actin | [1][3][14][9,11,22] |
| Actin-nucleating | Formin, Arp2/3 complex, proteins with tandem WH2 domains, leiomodin | Nucleation to initiate actin polymerization | [3][15][16][17][11,23,24,25] |
| Actin-elongating | Formin, tetramers of Ena/VASP | Regulation of actin assembly | [3][16][11,24] |
| Actin-bundling | Fimbrin/Plastin, hhLIM, gelsolin | Causes parallel F-actin filaments to closely pack together | [18][19][20][21][26,27,28,29] |
| Severing | ADF/cofilin, gelsolin, twinfilin, FRL-α, INF-2 | Severs F-actin | [22][23][24][25][26][30,31,32,33,34] |
| Capping | Twinfilin, gelsolin, tropomodulin, CapZ, Arp2/3 complex | Caps F-actin to inhibit actin polymerization | [3][27][28][29][11,35,36,37] |
| Motor | Myosin | Cargo transfer | [30][38] |
2. ABPs in Cardiac Hypertrophy
2.1. Profilin-1
Profilin is widely expressed in most eukaryotes and has a molecular weight of about 17 kDa [31][39]. There are various profilin isoforms expressed in different tissues. Profilin-1 is universally expressed, profilin-2 is specifically expressed in the brain and profilin-3 and profilin-4 are specifically expressed in kidney and testis, respectively [32][40]. Profilin accelerates the nucleotide exchange of G-actin and delivers ATP-G-actin to the growing barbed ends of F-actin through interacting with the poly-proline motifs of formin, vasodilator-stimulated phosphoprotein (VASP) and CDC42-activated Wiskott Aldrich syndrome protein (WASP)/WASP family [12][33][34][35][20,41,42,43]. Profilin-1 is directly associated with cardiac hypertrophy [36][44]. Overexpression of profilin-1 in the vascular tissues of FVB/N mice leads to vascular remodeling and hypertension by increasing actin aggregation, which provides mechanical stress for the development of cardiac hypertrophy [37][38][45,46]. It has been shown that the protein level of profilin-1 is significantly increased in mammalian hypertrophic hearts (Figure 12). The myocardin-related transcription factor megakaryoblastic leukemia (MKL) induces the expression of the signal transducer and activator of transcription 1 (STAT1) via its SAP-domain (SAF-A/B, acinus and PIAS protein domain) activity, which upregulates PFN expression [39][47]. Whether this is the explanation for the increased protein level of profilin in cardiac hypertrophy remains to be investigated. In cardiomyocytes, the functional abnormality of profilin-1 can change the abundance or activity of multiple proteins associated with cardiomyopathy. For example, the overexpression of profilin-1 can contribute to decreases in the phosphorylation level of endothelial nitric oxide synthases (eNOS) at Ser1177 in the hearts of spontaneous hypertensive rats [9][17]. Levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), skeletal muscle α-actin (α-SMA) and phosphorylated ERK1/2 (active form) were significantly increased in neonatal rat ventricular myocytes (NRVMs) following stimulation by phenylephrine or endothelin 1, which can be inhibited by siRNA-directed PFN1 silencing [36][44]. Increased phosphorylation of ERK1/2 activates the mechanistic (mammalian) target of rapamycin complex 1 (mTORC1) that subsequently inhibits autophagy [40][41][42][48,49,50]. It may be a potential key mechanism of cardiac hypertrophy mediated by the dysregulation of profilin-1 (Figure 12). Additionally, the inhibition of Rho-associated coiled-coil-containing protein kinase pathway (ROCK) can suppress the upregulation of profilin-1 induced by advanced glycation end products (AEGs) in H9c2 cells [43][51]. By comparison, overexpression of PFN1 results in the reactivation of fetal genes (NPPA and NPPB), an increase in F-actin in myocardium and destruction of myofibrils [36][44]. These processes can be reversed by inhibiting the expression of profilin-1 [9][17]. The inhibition of profilin-1 expression in H9c2 cells and Sprague–Dawley rats can attenuate cardiac hypertrophy induced by AEGs [43][44][51,52]. In Drosophila, myocyte-specific overexpression of profilin leads to disorders in muscle fibers and sarcomeres, which result in damaged muscle ultrastructure and function [36][44].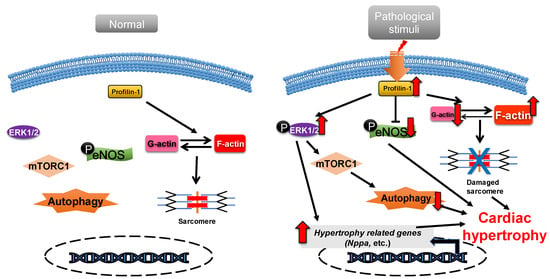
Figure 12. Profilin-1 mediates cardiac hypertrophy. In normal cardiomyocytes, profilin-1 is at a basal level and the fetal genes are not activated. Pathological stimuli increase the protein level of profilin-1, which results in ERK1/2 activation, F-actin accumulation and eNOS inhibition. This results in the reactivation of hypertrophy-related genes, inhibition of autophagy and damage to sarcomere structure and, ultimately, the development of cardiac hypertrophy.
2.2. ADF/Cofilin
Actin-depolymerizing factor (ADF)/cofilin consists of a single ADF homologous domain and has a molecular weight of about 15 kDa. The ADF/cofilin family contains ADF (also known as destrin, mainly expressed in endothelial and epithelial cells) and two cofilin isoforms (cofilin-1 is universal and cofilin-2 is cardio-specific) [45][46][53,54]. ADF/cofilin can bind to both G-actin and F-actin and can sever and depolymerize F-actin in regulating actin dynamics, which contributes to the cell contractility power [47][55]. The activity of cofilin is regulated by phosphorylation primarily from the ROCK/Lin-11, Isl1 and MEC-3 domain kinase (LIMK)/cofilin signaling pathway (Figure 23) [48][49][56,57]. Cofilin is inactivated via phosphorylation.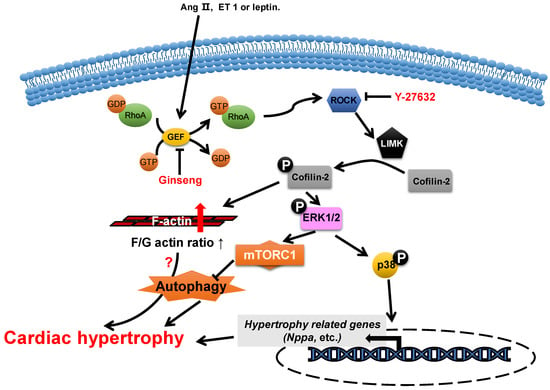
Figure 23. Proposed roles of cofilin-2 in cardiac hypertrophy. Neurohumoral factors (e.g., Ang II, ET 1 and leptin) lead to cofilin-2 phosphorylation through the RhoA/ROCK/LIMK signaling pathway. Phosphorylated cofilin-2 can lead to F-actin accumulation, which may subsequently contribute to cardiac hypertrophy through disrupting autophagy. In addition, it promotes the activation of ERK1/2 and p38, which contributes to the inhibition of autophagy and the reactivation of hypertrophy-related genes, which subsequently cause cardiac hypertrophy.
2.3. Formin
Formin is a type of multidomain protein consisting of 7 subfamilies and 15 members in human genes. Formins are characterized by the presence of two conserved domains: formin homology 1 (FH1) and FH2. FH1 binds to the profilin–actin complex via poly-proline sequences and brings the G-actin to FH2, which promotes actin nucleation and polymerization [3][57][11,65].2.4. CapZ
CapZ, a type of capping protein, anchors F-actin to the Z disc and regulates actin turnover, which contributes to sarcomere structural changes [58][59][76,77]. PIP2, a downstream effector of RAC1, can promote the dissociation of CapZ from F-actin by weakening their binding affinity [60][61][62][78,79,80]. Overexpression of CapZ in transgenic mice can lead to fatal cardiac hypertrophy [59][77]. It has been shown that hypertrophic agonists, phenylephrine or endothelin can reduce the binding affinity between CapZ and F-actin via PIP2-dependent pathways in NRVMs [63][81]. This may result in sarcomere remodeling, which induces cardiac hypertrophy. The cyclic mechanical strain activates downstream focal adhesion kinase (FAK) via the mechanotransduction of integrin, which then activates phosphatidylinositol 4-phosphate 5-kinase (PIP5K) through the RhoA/ROCK pathway. PIP5K phosphorylates phosphatidylinositol 4-phosphate (PI4P) in order to produce PIP2, which reduces the affinity of CapZ and F-actin binding, which contributes to the dysregulation of F-actin assembly and cardiac hypertrophy (Figure 35) [62][64][65][66][80,82,83,84].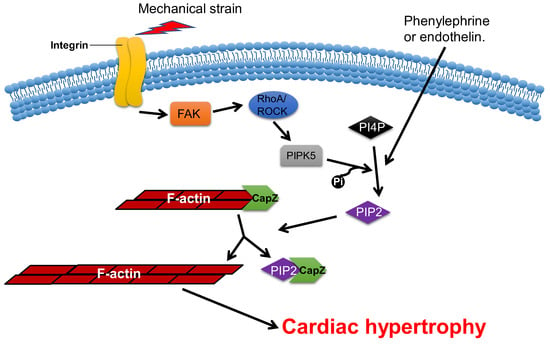
Figure 35. CapZ regulates cardiac hypertrophy. Mechanotransduction leads to the activation of RhoA/Rho-kinase pathway through integrins, which reduce the binding affinity of CapZ and F-actin. It subsequently causes cardiac hypertrophy.