Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 3 by Beatrix Zheng.
Recent studies have indicated a central role for LonP1 in mitochondrial function. Its physiological functions include proteolysis, acting as a molecular chaperone, binding mitochondrial DNA, and being involved in cellular respiration, cellular metabolism, and oxidative stress. Given its vital role in energy metabolism, LonP1 has been suggested to be associated with multi-system neoplasms and developmental disorders.
- LonP1
- mitochondrial Lon protease
- proteolysis
- molecular chaperone
1. Introduction
The mitochondrial Lon protease, alternatively called LonP1, is a protease that is encoded by the nucleus but resides in the mitochondrial matrix. Over 40 years ago [1], LonP1 was studied for its powerful physiological functions, including the degradation of oxidized or damaged proteins, modulation of mitochondrial DNA (mtDNA), and maintenance of mitochondrial homeostasis. These functions enable LonP1 to mediate vital physiological processes such as cellular respiration, cellular metabolism, and oxidative stress, but also to contribute to certain diseases, including neoplasms, developmental defects, neurodegenerative diseases, and cardiac disorders. Recently, LonP1 has been found to play an essential role in the growth of and tumorigenesis in oral and maxillofacial regions. For example, patients with cerebral, ocular, dental, auricular, and skeletal syndrome (CODAS) with mutations in the LONP1 gene exhibit unique oral and maxillofacial features, including a broad, flattened skull, collapsed midface, delayed tooth eruption, and abnormally-shaped teeth [2]. LonP1 has also been found to be markedly overexpressed in tissue samples derived from oral, head, and neck cancers, thereby contributing to their invasion, metastasis, angiogenesis, and resistance to treatment through multiple pathways [3].
2. LonP1 in the Oral and Maxillofacial Regions
2.1. LonP1 in Oral and Maxillofacial Tumorigenesis
2.1.1. Roles of LonP1 in Oral and Maxillofacial Tumorigenesis
LonP1 appears to be upregulated in various neoplasms. LonP1 has been found to be upregulated in head and neck cancer tissues, ranking in the top 1% in silico. In the laboratory, oral cancer-derived cells and tissue samples from oral squamous cell carcinoma (OSCC) showed a significant upregulation of LonP1 compared to cells or tissues without tumors [3]. In addition to oral and head and neck cancers, LonP1 was also found to be overexpressed in cells and tissue samples from malignant lymphoma [4], cervical neoplasms [5], lung adenocarcinoma, lung cancer, malignant melanoma, breast cancer [3], bladder tumors [6], leukemia [7], glioma [8], colorectal cancer [9] and renal cell carcinoma [10].
Elevated LonP1 expression is correlated with progressive TNM staging, severe histological grading, and high cancer aggressiveness. As an independent prognostic factor for cancer, high levels of LonP1 are correlated with low survival [6][9]. In vitro, LonP1 overexpression promotes the survival, proliferation, and migration of cancer cells, thereby triggering epithelial-to-mesenchymal transition [3]. In contrast, LonP1-deficient cancer cells exhibit attenuated proliferation, increased apoptosis, decreased cellular bioenergy, and reduced drug resistance. LonP1 deficiency protects mice from chemically-induced colorectal cancer. Furthermore, triterpenoids that irreversibly suppress LonP1 activity have been found to induce apoptosis and inhibit cancer cell proliferation [11].
2.1.2. Mechanisms of LonP1 in Oral and Maxillofacial Tumorigenesis
The mechanisms underlying LonP1 carcinogenesis are not yet fully understood. Several studies have shown that the mechanisms by which LonP1 promotes oral and maxillo-facial cancer are complicated, with a key step being the upregulation of ROS. ROS are closely associated with cancer [12]. They promote the induction of nuclear DNA and mtDNA lesions and contribute to cancer [13]. ROS also serve as signaling molecules that stimulate cancer cell migration and invasion and promote cancer-associated fibroblast formation in the tumor microenvironment, thereby boosting cancer growth and aggressiveness [12]. ROS even promote tumor angiogenesis. In oral cancer-derived cell lines (OEC-M1 and FADU), the upregulation of LonP1 triggers ROS production, which further promotes proliferation via Ras-ERK signaling and migration and invasion via the ERK and p38 MAPK pathways [3]. In addition, the LonP1-ROS axis stimulates inflammatory factor generation and triggers epithelial-to-mesenchymal transition, angiogenesis, and M2 macrophage polarization, thereby leading to the growth of an immunosuppressive tumor microenvironment that encourages cancer development and metastasis [14]. The LonP1-ROS axis also causes the release of mtDNA, which results in PD-L1-mediated immune escape via the interferon pathway and extracellular vesicles. Therefore, in oral cancer, mtDNA and PD-L1 in extracellular vesicles may serve as promising markers for anti-PD-L1 immunotherapy [15]. LonP1 also regulates the electron transport chain complex of the OXPHOS, NDUFS8, which modulates ROS production and promotes proliferation and invasion [16]. In addition to the aforementioned findings, other research teams have confirmed the involvement of the LonP1-ROS axis in promoting other types of cancers [17][18].
LonP1 further regulates carcinogenesis by modulating the bioenergetics of tumor cells. The knockdown of LonP1 in bladder cancer, cervical cancer, prostate adenocarcinoma, and glioma cells induces a decline in mitochondrial respiratory function with reduced ATP production [5][6][17][19], and some cells exhibit the downregulation of both OXPHOS and glycolytic pathways [5][6]. Cancer cells that are deficient in LonP1 have been found to exhibit a decreased capacity to proliferate. LonP1 has also been reported to be involved in the metabolic conversion of OXPHOS to glycolysis. This is a key feature of malignancy. Cells with oncogenic phenotypes convert their metabolic pattern from aerobic OXPHOS to anaerobic glycolysis due to the rapid glycolytic turnover, faster and greater ATP production, and availability of a large number of metabolic intermediates to accommodate rapid cell proliferation and large-scale biosynthesis [20]. During the carcinogenesis of gastric epithelial cells infected with H. pylori, LonP1 overexpression has been found to promote the conversion of metabolic patterns to glycolysis and the massive proliferation of cells. In contrast, LonP1 knockdown significantly attenuates glycolysis and proliferation.
LonP1 has also been found to boost vimentin and N-calmodulin levels, while E-calmodulin levels decline, implying that LONP1 may be involved in epithelial-to-mesenchymal transition [3]. The molecular chaperone activity of LonP1 is also involved in the emergence and progression of oral cancers. In oral cancer cells, LonP1 suppresses p53-dependent apoptosis under oxidative stress by stabilizing and binding to p53. LonP1 levels have been shown to correlate with cytoplasmic P53 levels in OSCC samples [21]. Recent studies have found that LonP1 affects the cancer phenotype by regulating the intracellular transport of mitochondria [17], and cell resistance to hypoxia [19].
In summary, the mechanisms by which LonP1 causes oral and maxillofacial cancers may include ROS generation, disturbed energy metabolism, the upregulation of glycolysis, resistance to hypoxia, mtDNA leakage, and mitochondrial subcellular transport.
2.1.3. LonP1 as a Target for Anticancer Therapy
LonP1 plays an important role in oncogenesis, cancer progression, and cancer cell invasion. LonP1-deficient cancer cells exhibit attenuated cell proliferation, increased apoptosis, and reduced drug resistance in vitro. LonP1 deficiency has been found to protect mice from chemically-induced colorectal cancer in vivo. Therefore, the establishment of LonP1 inhibitors may be a promising antitumor chemotherapy approach.
In 2008, Bayot et al. proposed coumarinic derivatives as inhibitors of LonP1 but did not study their effects on tumors [22]. In 2010, Wang et al. found that obtusilactone A and (-)-sesamin promoted cell death in lung carcinoma through the targeted suppression of LonP1 [23]. Subsequently, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) and its derivatives were found to contribute to lymphoma cell death by downregulating LonP1 [4]. In colon, liver, and breast cancer cells, CDDO induces depolarization, increases mitochondrial ROS, and alters mitochondrial morphology by targeting LonP1, thereby promoting apoptosis in cancer cells [11].
Recently, the LonP1-NCLX axis has been found to promote cisplatin resistance in oral cancer cells. An association has also been noted between NCLX and LonP1 levels in oral cancer tissue samples. Inhibitors of LonP1 and NCLX may be used as potential adjuvants to cisplatin therapy to avoid chemoresistance and enhance the curative effect in patients with oral cancer [24].
2.2. LonP1 in Oral and Maxillofacial Development
2.2.1. Roles of LonP1 in Oral and Maxillofacial Development
In 1991, Shehib et al. [25] reported a unique case with clinical manifestations comprising delayed brain development, cataracts, flattened craniofacial bones, a fluted nasal tip, delayed tooth eruption and its abnormal morphology, unusual ear shape or compromising hearing, short stature, and delayed epiphyseal development. This condition became known as cerebral, ocular, dental, auricular, and skeletal syndrome (CODAS; MIM 600373). To date, CODAS cases remain rare, with no more than 20 reported cases worldwide. Interestingly, in 2015, two separate studies were published by two large research teams almost simultaneously. Sanger sequencing confirmed that the reported patients with CODAS had mutations in the LONP1 gene [2][26].
The first reported case of CODAS included detailed dental information [25]. The little girl did not present with the partial eruption of her deciduous second molar teeth until she was 40 months old, whereas the eruption of other primary teeth occurred when she was between 24 and 30 months old. The occlusal surfaces of her deciduous canine and the first and second deciduous molars had an abnormal morphology, such as sunken dishes, which were prone to the retention of dental plaque. Anomalous extended cusps resulted in an open bite and multiple ulcers on the ventral and lateral sides of the tongue. Unfortunately, owing to the lack of facilities available at that time, only a written description and black-and-white photographic documentation of the dental symptoms of the patient were made (see Shebib SM, Am J Med Genet. 1991 [25] for details of the photographic records). For this patient, in 2015, a homozygous mutation in the LONP1 gene (LONP1 c.2026C > T [p.Pro676Ser]) was identified, which is located adjacent to the ATP binding pocket and may compromise LonP1’s proteolytic and ATP hydrolysis capabilities (Table 1) [2].
Table 1. Mutation details of the LONP1 gene in CODAS patients with dental abnormalities.
| Publication | Patients Amount | Homozygous/hterozygous | Mutation Type |
Mutation Location |
LONP1 Variants |
|---|---|---|---|---|---|
| Strauss KA, Am J Hum Genet. 2015 | 1 patient | Homozygous | Unclear | Adjacent to the ATP binding pocket | LONP1 c.2026C > T [p.Pro676Ser] |
| Strauss KA, Am J Hum Genet. 2015 | 8 patients | Homozygous | Unclear | AAA+ module | LONP1 c.2161C > G [p.Arg721Gly] |
| Strauss KA, Am J Hum Genet. 2015 | 1 patient | Heterozygous | Unclear | AAA+ module | LONP1 c.1892C > A/c.2171C > T [p.Ser631Tyr /p.Ala724Val] |
| Dikoglu E, Am J Med Genet A. 2015 | 1 patient | Heterozygous | Missense mutation & shift mutation | Truncated LonP1 and deletion of ATPase and P domains | c.1392G > A p.W464 * |
* Represents protein truncation.
In addition to assessing the Sanger sequencing results from the first case, Strauss et al. [2] collected data from nine other patients with CODAS for sequencing. All of the cases presented with delayed tooth eruption and abnormal tooth morphology; however, de-tailed medical records and intraoral photographs were not available. Eight of the cases were homozygous (LONP1 c.2161C > G [p.Arg721Gly]) and one was compound heterozygous (LONP1 c.1892C > A/c.2171C > T [p.Ser631Tyr /p.Ala724Val]). All of the mutations were located in the AAA+ module responsible for ATP binding, which led to reduced ATP hydrolysis and impaired proteolysis (Table 1) [27]. The lymphoblastoid cell line from the patients (p.Arg721Gly/p.Arg721Gly) exhibited enlarged mitochondria with electron-dense inclusion bodies and anomalous morphology, a significant decrease in mtDNA-encoded cytochrome c oxidase subunit II, and an impaired alternate respiratory capacity, which contributed to compromised mitochondrial homeostasis and function.
Dikoglu et al. [26] studied seven patients with CODAS and performed Sanger sequencing on their data. Only one patient had dental manifestations (delayed tooth eruption and cusp elongation). Unfortunately, the study only showed orthopedic radiographs and clinical photographs of the head and face of the patient but no dental photographs, which was likely due to the young age of the patient, who could not easily cooperate. The patient was heterozygous for a missense mutation and shift mutation in the LONP1 gene, which resulted in a truncated LonP1 protein and deletion of the ATPase and protease domains (Table 1). Other patients with no dental manifestations were heterozygous or homozygous for missense mutations in LONP1 and only exhibited mental retardation, cataracts, and epiphyseal dysplasia with or without hearing impairment (Figure 1).
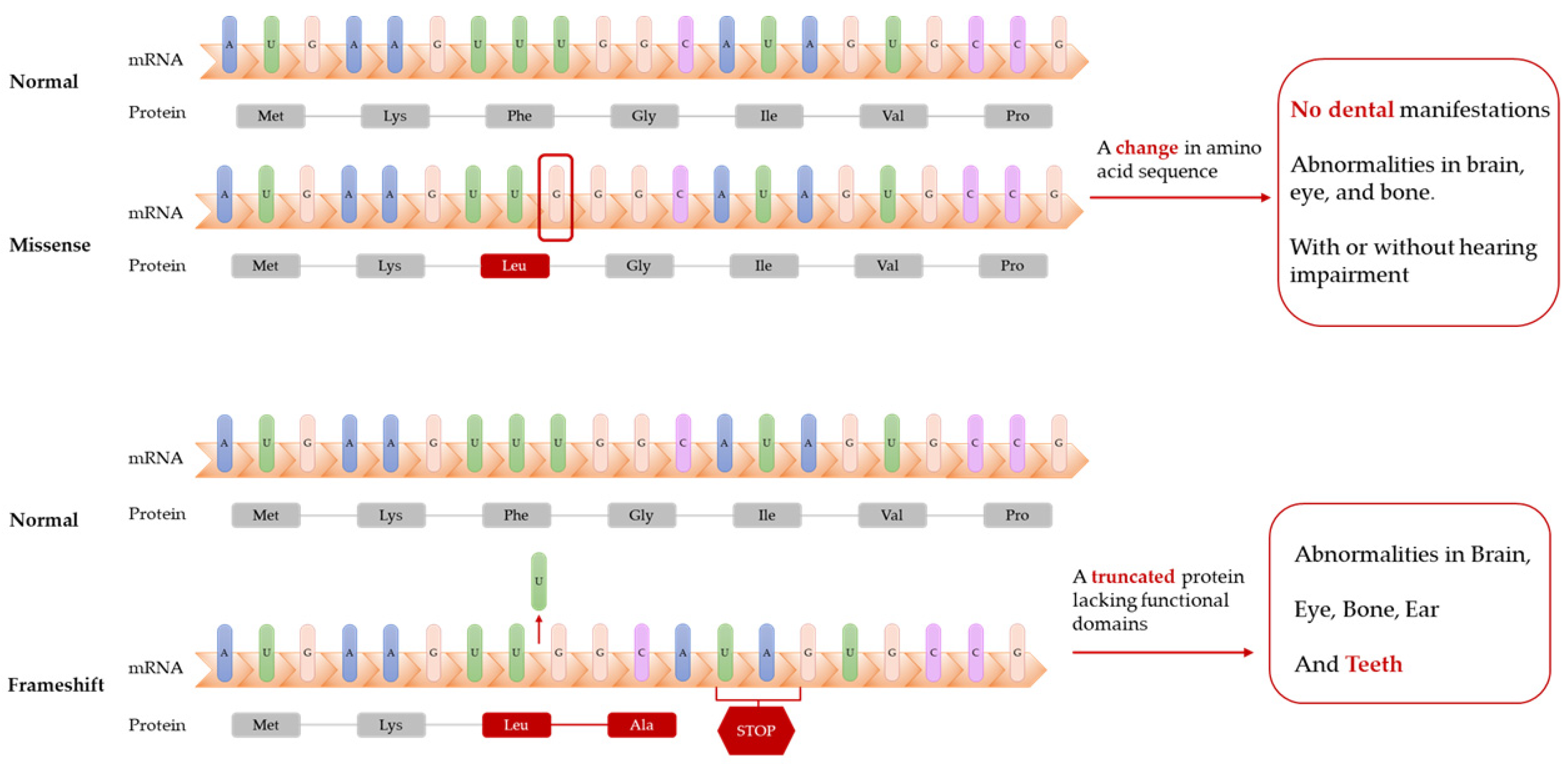
Figure 1. Frameshift mutations of LONP1 gene lead to truncated proteins. Patients with these mutations have dental abnormalities (delayed tooth eruption and cusp elongation), while patients with missense mutations of LONP1 gene might not have dental manifestations.
The abovementioned studies suggest that biallelic mutations in LONP1 comprise the etiology of CODAS (Table 1). Mutations occurring in the AAA+ module and shift mutations are most likely to contribute to delayed dental development and morphological abnormalities of the teeth, whereas other mutations may only cause developmental abnormalities in the brain, eyes, and bones.
2.2.2. Possible Mechanisms of Action of LonP1 in Oral and Maxillofacial Development
The mechanisms by which LonP1 affects oral and maxillofacial tissue development and cell differentiation are unclear. However, insights may be gained from studies on the mechanisms by which LonP1 regulates the development of other tissues or cell differentiation. The most likely mechanism involves the LonP1-driven regulation of development through energy metabolism [28][29][30]. For example, there is a critical time point in murine embryonic heart development, E11.5, when cardiac metabolism is converted from anaerobic glycolysis to OXPHOS [28]. However, murine hearts with a deletion of LonP1 still maintain high glycolytic activity at E12.5, and myocardial development is impaired until the mice die of heart failure at birth [28]. It is believed that LonP1 is indispensable for cardiomyocytes to complete metabolic conversion, which would otherwise impair cardiac development. LonP1 is essential for myoblast myogenic differentiation. A deficiency of LonP1 inhibits myoblast mitophagy, suppresses mitofission and mitofusion, leads to mitochondrial depolarization, and thus, fails to complete differentiation [30].
References
- Waxman, L.; Goldberg, A.L. Protease La from Escherichia coli hydrolyzes ATP and proteins in a linked fashion. Proc. Natl. Acad. Sci. USA 1982, 79, 4883–4887.
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135.
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681.
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329.
- Nie, X.; Li, M.; Lu, B.; Zhang, Y.; Lan, L.; Chen, L.; Lu, J. Down-regulating overexpressed human Lon in cervical cancer suppresses cell proliferation and bioenergetics. PLoS ONE 2013, 8, e81084.
- Liu, Y.; Lan, L.; Huang, K.; Wang, R.; Xu, C.; Shi, Y.; Wu, X.; Wu, Z.; Zhang, J.; Chen, L.; et al. Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: Potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget 2014, 5, 11209–11224.
- Goto, M.; Miwa, H.; Suganuma, K.; Tsunekawa-Imai, N.; Shikami, M.; Mizutani, M.; Mizuno, S.; Hanamura, I.; Nitta, M. Adaptation of leukemia cells to hypoxic condition through switching the energy metabolism or avoiding the oxidative stress. BMC Cancer 2014, 14, 76.
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200.
- Wang, B.; Yin, X.; Gan, W.; Pan, F.; Li, S.; Xiang, Z.; Han, X.; Li, D. PRCC-TFE3 fusion-mediated PRKN/parkin-dependent mitophagy promotes cell survival and proliferation in PRCC-TFE3 translocation renal cell carcinoma. Autophagy 2021, 17, 2475–2493.
- Gibellini, L.; Pinti, M.; Bartolomeo, R.; De Biasi, S.; Cormio, A.; Musicco, C.; Carnevale, G.; Pecorini, S.; Nasi, M.; De Pol, A.; et al. Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget 2015, 6, 25466–25483.
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297.
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721.
- Kuo, C.L.; Chou, H.Y.; Chiu, Y.C.; Cheng, A.N.; Fan, C.C.; Chang, Y.N.; Chen, C.H.; Jiang, S.S.; Chen, N.J.; Lee, A.Y. Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. 2020, 474, 138–150.
- Cheng, A.N.; Cheng, L.C.; Kuo, C.L.; Lo, Y.K.; Chou, H.Y.; Chen, C.H.; Wang, Y.H.; Chuang, T.H.; Cheng, S.J.; Lee, A.Y. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer 2020, 8, 1372.
- Bota, D.A.; Davies, K.J.A. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680.
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 2019, 38, 6926–6939.
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Lo Tartaro, D.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 Differently Modulates Mitochondrial Function and Bioenergetics of Primary Versus Metastatic Colon Cancer Cells. Front. Oncol. 2018, 8, 254.
- Di, K.; Lomeli, N.; Wood, S.D.; Vanderwal, C.D.; Bota, D.A. Mitochondrial Lon is over-expressed in high-grade gliomas, and mediates hypoxic adaptation: Potential role of Lon as a therapeutic target in glioma. Oncotarget 2016, 7, 77457–77467.
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152.
- Sung, Y.J.; Kao, T.Y.; Kuo, C.L.; Fan, C.C.; Cheng, A.N.; Fang, W.C.; Chou, H.Y.; Lo, Y.K.; Chen, C.H.; Jiang, S.S.; et al. Mitochondrial Lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. 2018, 9, 697.
- Bayot, A.; Basse, N.; Lee, I.; Gareil, M.; Pirotte, B.; Bulteau, A.-L.; Friguet, B.; Reboud-Ravaux, M. Towards the control of intracellular protein turnover: Mitochondrial Lon protease inhibitors versus proteasome inhibitors. Biochimie 2008, 90, 260–269.
- Wang, H.-M.; Cheng, K.-C.; Lin, C.-J.; Hsu, S.-W.; Fang, W.-C.; Hsu, T.-F.; Chiu, C.-C.; Chang, H.-W.; Hsu, C.-H.; Lee, A.Y.-L. Obtusilactone A and (−)-sesamin induce apoptosis in human lung cancer cells by inhibiting mitochondrial Lon protease and activating DNA damage checkpoints. Cancer Sci. 2010, 101, 2612–2620.
- Tangeda, V.; Lo, Y.K.; Babuharisankar, A.P.; Chou, H.-Y.; Kuo, C.-L.; Kao, Y.-H.; Lee, A.Y.-L.; Chang, J.-Y. Lon upregulation contributes to cisplatin resistance by triggering NCLX-mediated mitochondrial Ca2+ release in cancer cells. Cell Death Dis. 2022, 13, 241.
- Shebib, S.M.; Reed, M.H.; Shuckett, E.P.; Cross, H.G.; Perry, J.B.; Chudley, A.E. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome--a case report. Am. J. Med. Genet. 1991, 40, 88–93.
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A 2015, 167, 1501–1509.
- Sha, Z.; Montano, M.M.; Rochon, K.; Mears, J.A.; Deredge, D.; Wintrode, P.; Szweda, L.; Mikita, N.; Lee, I. A structure and function relationship study to identify the impact of the R721G mutation in the human mitochondrial lon protease. Arch. Biochem. Biophys. 2021, 710, 108983.
- Zhao, K.; Huang, X.; Zhao, W.; Lu, B.; Yang, Z. LONP1-mediated mitochondrial quality control safeguards metabolic shifts in heart development. Development 2022, 149, dev200458.
- Venkatesh, S.; Baljinnyam, E.; Tong, M.; Kashihara, T.; Yan, L.; Liu, T.; Li, H.; Xie, L.H.; Nakamura, M.; Oka, S.I.; et al. Proteomic analysis of mitochondrial biogenesis in cardiomyocytes differentiated from human induced pluripotent stem cells. Am. J. Physiol. Regul. Integr. Comp. physiol. 2021, 320, R547–R562.
- Huang, S.; Wang, X.; Yu, J.; Tian, Y.; Yang, C.; Chen, Y.; Chen, H.; Ge, H. LonP1 regulates mitochondrial network remodeling through the PINK1/Parkin pathway during myoblast differentiation. Am. J. Physiol. Cell Physiol. 2020, 319, C1020–C1028.
More