Glaucoma is a progressive age-related disease of the visual system and the leading cause of irreversible blindness worldwide. Currently, intraocular pressure (IOP) is the only modifiable risk factor for the disease, but even as IOP is lowered, the pathology of the disease often progresses. Hence, effective clinical targets for the treatment of glaucoma remain elusive. Glaucoma shares comorbidities with a multitude of vascular diseases, and evidence in humans and animal models demonstrates an association between vascular dysfunction of the retina and glaucoma pathology. Integral to the survival of retinal ganglion cells (RGCs) is functional neurovascular coupling (NVC), providing RGCs with metabolic support in response to neuronal activity. NVC is mediated by cells of the neurovascular unit (NVU), which include vascular cells, glial cells, and neurons. Nitric oxide-cyclic guanosine monophosphate (NO-cGMP) signaling is a prime mediator of NVC between endothelial cells and neurons, but emerging evidence suggests that cGMP signaling is also important in the physiology of other cells of the NVU. NO-cGMP signaling has been implicated in glaucomatous neurodegeneration in humans and mice.
- cGMP
- glaucoma
- neurovascular coupling
- neurodegeneration
- glia
- endothelial cell
- retina
- neurovascular unit
1. Introduction
2. The Retinal Neurovascular Unit and Glaucoma
The visual system has developed intricate regulatory mechanisms to fulfill the high metabolic demand of the retina. One such mechanism is neurovascular coupling (NVC), whereby an increased neuronal activity promotes signaling between cells of the neurovascular unit (NVU), resulting in increased supply of metabolites and removal of waste products by the vasculature [12][7]. The retinal NVU consists of three main cell types: neurons, glial cells (including astrocytes, microglia, and oligodendrocytes), and vascular cells (endothelial cells, vascular smooth muscle cells, and pericytes; Figure 1) [13][8]. Dysfunction in the NVU is evident in the pathophysiology of many neurodegenerative diseases of the central nervous system (CNS), including Alzheimer’s Disease and Parkinson’s Disease [3]. Such neurodegenerative diseases share many common pathophysiologies with glaucoma, and thus dysfunction in the NVU may also be a relevant mechanism of neurodegeneration of the visual system [3,13][3][8].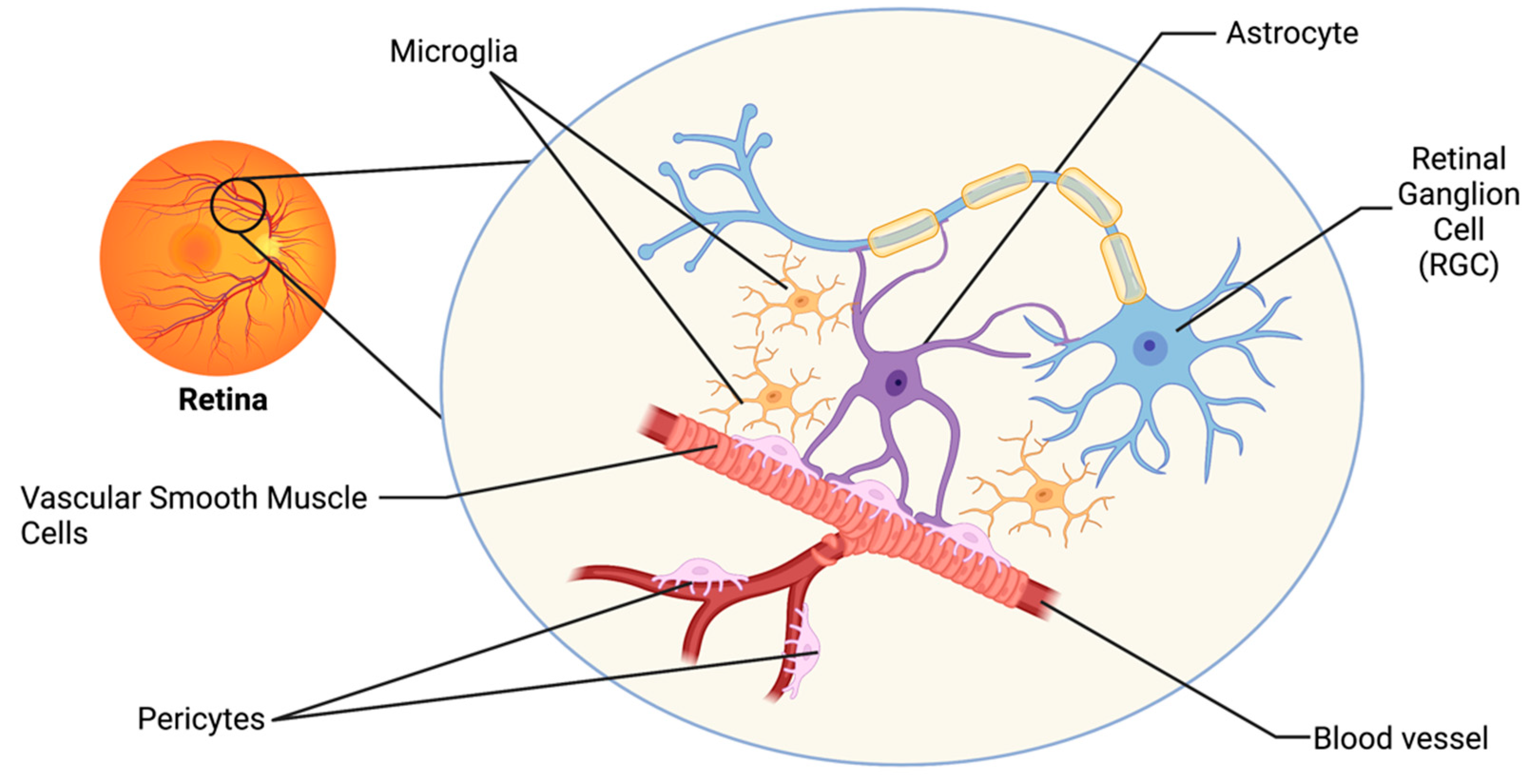
3. NO-cGMP Signaling in Glaucoma
The nitric oxide-cyclic guanosine monophosphate (NO-cGMP) signaling pathway is integral to NVC mechanisms and is the prime mediator of vasodilation in endothelial cells [9][11]. NO is produced by nitric oxide synthase enzymes in cells of the NVU in both physiological and pathophysiological conditions [17][12]. NO is lipophilic and can freely traverse cellular membranes, where it binds to its primary receptor, soluble guanylate cyclase (GC1; [18,19][13][14]). GC1 binding of NO stimulates the production of cGMP, a second messenger capable of binding to multiple cellular protein targets such as cyclic nucleotide phosphodiesterases (PDEs), cGMP-dependent protein kinases, and cGMP-gated ion channels [20,21,22,23][15][16][17][18]. cGMP signaling is multifaceted, with the potential to exert broad effects on cellular physiology, depending upon the cellular target that it binds. Dysfunction in cGMP signaling has been implicated in glaucoma pathophysiology in human patients and in animal models of the disease. Levels of NO and cGMP in aqueous humor and plasma are reduced in glaucoma patients [24,25][19][20]. Furthermore, in a genome-wide association study (GWAS) of patients with primary open angle glaucoma (POAG), a single nucleotide polymorphism (rs11722059) located in the GUCY1A3/GUCY1B3 gene locus was significantly associated with paracentral visual field loss in female glaucoma patients [26][21]. GUCY1A3/GUCY1B3 encode the alpha and beta subunits of GC1, respectively [26][21]. Identification of a risk variant in the GC1 gene and expression of GC1 in human and rodent ocular tissues including RGCs [26][21] suggests that GC1 may have a role in the pathogenesis of glaucoma.4. NO-cGMP Signaling in Vascular Cells
The vascular cells of the NVU in the brain consist of the endothelial cells, vascular smooth muscle cells, and pericytes. Nestled between the blood vessel lumen and the smooth muscle cells, the endothelium is responsible for modulation of vascular tone, thrombus formation, cell adhesion, smooth muscle proliferation, and sequestration of inflammatory factors [33][22]. Within the retinal NVU, the function is similar; endothelial cells line retinal vessels responsible for RGC blood supply and smooth muscle cells control vascular tone [9,34][11][23]. Critical to endothelial function is cGMP signaling; outside of the CNS, cGMP signaling plays a major role in decreasing endothelial cell permeability and is responsible for smooth muscle cell relaxation and subsequent vasodilation [35,36][24][25]. Within the NVU, NO-cGMP signaling plays a similar role in modulating vascular tone; NO produced in the endothelium acts on GC1 in smooth muscle cells to induce vasodilation via smooth muscle relaxation (Figure 2) [34][23]. Moreover, cGMP is also implicated in decreasing the permeability of the endothelium of the NVU. Experimental treatment with the cGMP agonist 8-Br-cGMP decreased the permeability of an in vitro blood–brain barrier (BBB) model [37][26].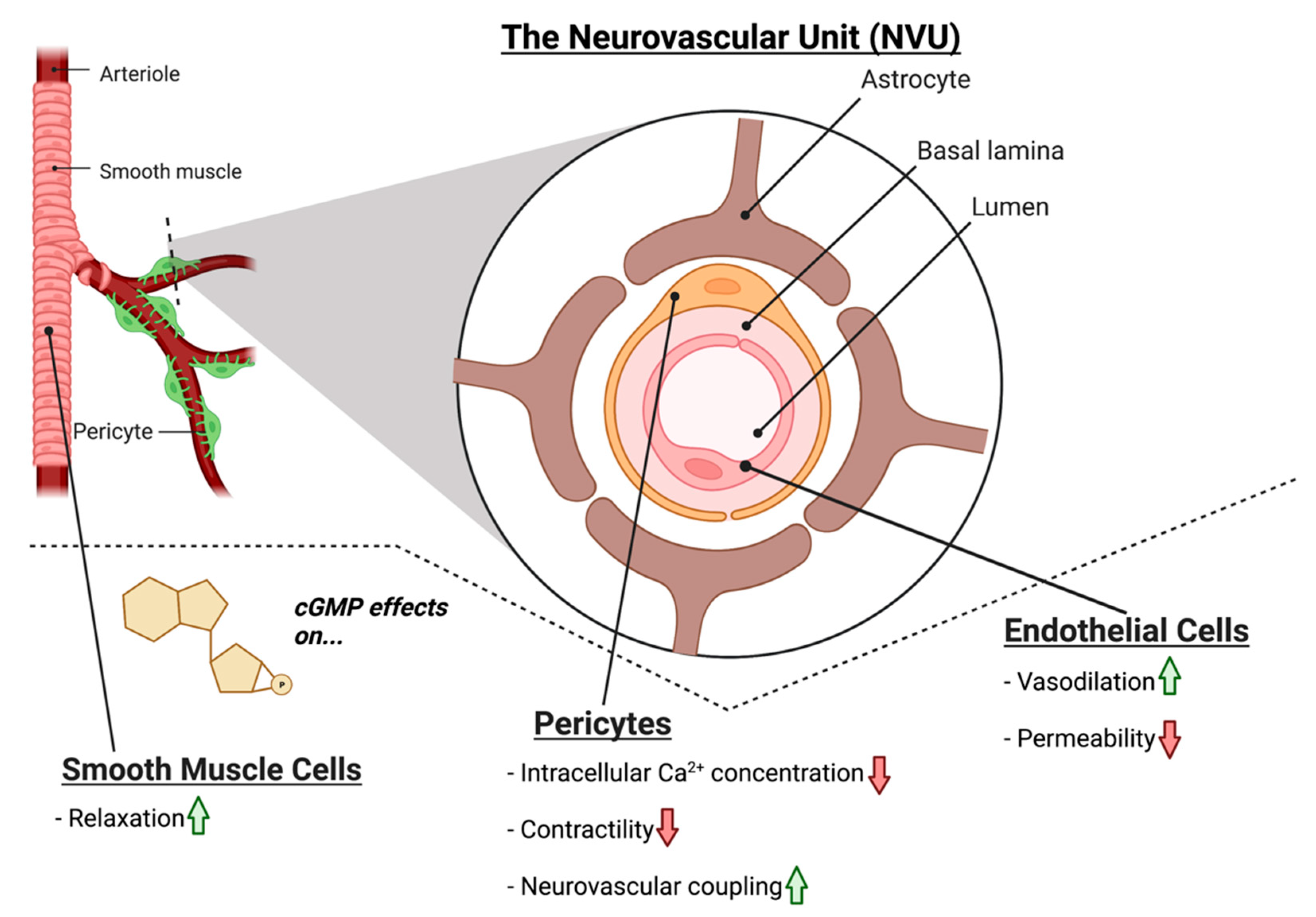
5. NO-cGMP Signaling in Glial Cells
The retinal glia, including astrocytes, Müller glia, and microglia, were thought to be relatively passive elements in the CNS; however, recently, it has become clear that they play a critical intermediary role between the neurons and vascular cells of the NVU [13,61,62][8][36][37]. The predominant glial cells in the retina are the Müller glia (MG), which span the retinal layers, from the inner border with the vitreous humor, to the photoreceptor layer. Astrocytes are present in the superficial layer of the retina and make intimate connections between RGCs and blood vessels. Astrocytes and MG together form the blood–retinal barrier (BRB) and are integral to retinal NVC [13,63,64][8][38][39]. Astrocytes play a critical role in shuttling energy resources to neurons, responding to glutamatergic activity by producing lactate and releasing it into the extracellular space, which neurons then utilize for metabolic demands [65,66,67][40][41][42]. Astrocytes also regulate neurotransmitter release and re-uptake, to help maintain neuronal homeostasis [13,68,69,70,71][8][43][44][45][46]. In humans, regarding glial cells, the fovea contains only MG subtypes, indicating that these cells alone are sufficient to supply the metabolic needs of the RGCs [13,71][8][46]. Microglia are also resident glial cells that span multiple layers of the retina, yet their exact role in the basal NVU function of the retina is less understood compared to other glial cell types [72][47]. In glaucoma, glial dysfunction and/or activation may contribute to breakdown in NVC, rendering RGCs more susceptible to degeneration. Given their localization in the RGC layer and role in metabolic support, changes in astrocyte physiology may have a large impact on the health and physiology of RGCs. During pathological conditions in the retina, astrocytes respond robustly with gross morphological and physiological changes, which may prompt the dysfunction and degeneration of RGC axons [9,67,74,75][11][42][48][49]. Examining the role of NO-cGMP signaling in the NVU may help to elucidate the relationship between glia, vascular function, and glaucoma pathology. In the CNS, decreased GC1 expression is detected in the reactive glia of post-mortem patients with neurodegenerative diseases such Alzheimer’s disease and multiple sclerosis [84,85,86][50][51][52]. In cultured retinal MG, addition of cGMP analogues activates a calcium permeable, non-selective cation current via the opening of cGMP-gated channels, indicating that cGMP signaling, in part, regulates MG physiology [87][53]. In the microglia, cGMP signaling pathways are involved in inflammatory gene expression [84,88,89,90,91,92,93,94][50][54][55][56][57][58][59][60]. Pharmacological inhibition of GC1 prevents activation and migration of microglia to injured or inflamed sites in the CNS; recruitment can be rescued with the addition of a cGMP analog, suggesting that microglial recruitment is, at least in part, mediated by cGMP signaling [95,96][61][62]. To date, the role of NO-cGMP signaling in astrocyte function has been explored in many contexts, both within the CNS and systemically, and has been summarized in Figure 3. In cultured rat cerebellar astrocytes, cGMP directly affects the motility and morphology of cells and expression of glial fibrillary acidic protein (GFAP), suggesting that cGMP signaling directly modulates astrocyte physiology [97,98][63][64]. In focal brain injury, inhibiting PDE increased cGMP and altered the glial inflammatory response, decreased oxidative stress and cell death, and increased angiogenesis (Figure 3) [84][50].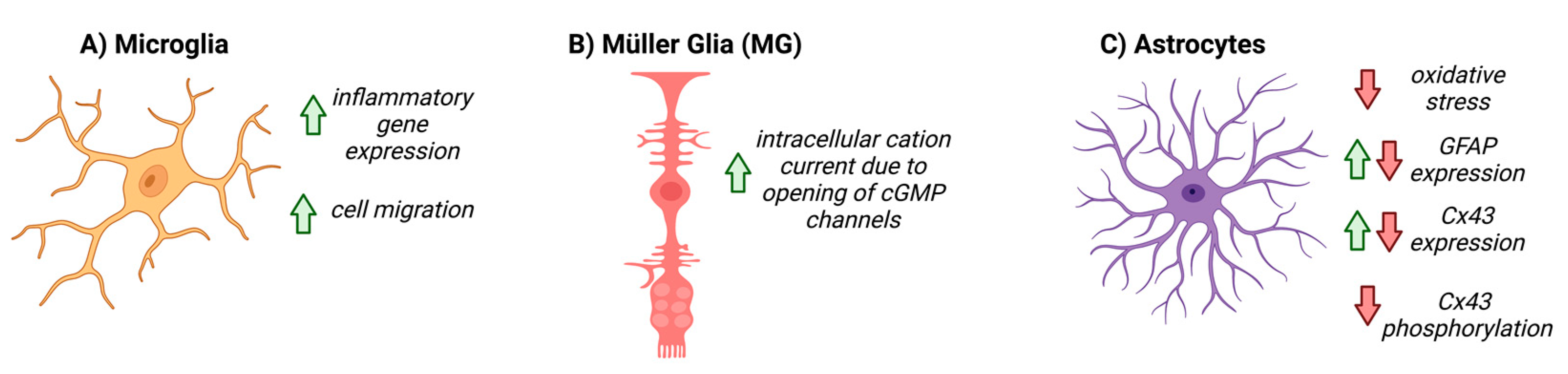
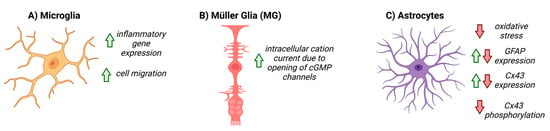
6. NO-cGMP Signaling in Retinal Neurons
In glaucoma, the degeneration of RGCs and their axons in the optic projection results in irreversible vision loss. The role of NO-cGMP signaling in RGCs appears to be multifaceted. In CNS neurons, NO-cGMP signaling is important in NVC, synaptic plasticity, and synaptic transmission [107][71]. GC1 is expressed in pre- and post-synaptic terminals and in neuronal soma and spines [107,108][71][72]. GC1 is also evident in RGC soma in the retina [26][21]. The depolarization of neuronal membranes stimulates Ca2+ influx into RGCs, which activates neuronal nitric oxide synthase, leading to NO production and release, stimulating vasodilation in its target vascular cells via cGMP [62][37]. Interestingly, GC1−/− mice [26][21] develop RGC degeneration with age, and increasing availability of cGMP has a neuroprotective effect in murine glaucoma models [28][73]. Furthermore, primary RGC cultures and mouse retinal explants exhibit decreased apoptotic signaling when supplemented with cGMP [109][74]. The transcription factor CREB is responsible for neuroprotective functions and is phosphorylated and activated by PKG in CNS neurons [110,119,120,121][75][76][77][78]. Alternatively, cGMP, via PKG, can also modulate intracellular calcium signaling in RGCs [122][79] and therefore may be responsible for limiting the neurotoxic effects to neurons of high intracellular Ca2+ levels. Since cGMP has multiple downstream effectors that may contribute to the neuroprotective effects observed in RGCs, further research is needed to distinguish the apparent neuroprotective and neurodegenerative arms of the cGMP signaling cascade.
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090.
- Calkins, D.J. Adaptive responses to neurodegenerative stress in glaucoma. Prog. Retin. Eye Res. 2021, 84, 100953.
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23.
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911.
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719.
- Chen, Y.Y.; Lai, Y.J.; Yen, Y.F.; Shen, Y.C.; Wang, C.Y.; Liang, C.Y.; Lin, K.H.; Fan, L.W. Association between normal tension glaucoma and the risk of Alzheimer’s disease: A nationwide population-based cohort study in Taiwan. BMJ Open 2018, 8, e022987.
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243.
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 9, 2641.
- Garhofer, G.; Zawinka, C.; Resch, H.; Huemer, K.H.; Schmetterer, L.; Dorner, G.T. Response of retinal vessel diameters to flicker stimulation in patients with early open angle glaucoma. J. Glaucoma 2004, 13, 340–344.
- Garhofer, G.; Resch, H.; Weigert, G.; Lung, S.; Simader, C.; Schmetterer, L. Short-term increase of intraocular pressure does not alter the response of retinal and optic nerve head blood flow to flicker stimulation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1721–1725.
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452.
- Dawson, T.M.; Dawson, V.L. Chapter Four—Nitric Oxide Signaling in Neurodegeneration and Cell Death. Adv. Pharmacol. 2018, 82, 57–83.
- Knowles, R.G.; Palacios, M.; Palmer, R.M.; Moncada, S. Formation of nitric oxide from L-arginine in the central nervous system: A transduction mechanism for stimulation of the soluble guanylate cyclase. Proc. Natl. Acad. Sci. USA 1989, 86, 5159–5162.
- Mergia, E.; Russwurm, M.; Zoidl, G.; Koesling, D. Major occurrence of the new alpha(2)beta(1) isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 2003, 15, 189–195.
- Sharina, I.G.; Jelen, F.; Bogatenkova, E.P.; Thomas, A.; Martin, E.; Murad, F. Alpha1 soluble guanylyl cyclase (sGC) splice forms as potential regulators of human sGC activity. J. Biol. Chem. 2008, 283, 15104–15113.
- Zagotta, W.N.; Siegelbaum, S.A. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 1996, 19, 235–263.
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563.
- Friebe, A.; Sandner, P.; Schmidtko, A. cGMP: A unique 2nd messenger molecule—Recent developments in cGMP research and development. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 287–302.
- Galassi, F.; Renieri, G.; Sodi, A.; Ucci, F.; Vannozzi, L.; Masini, E. Nitric oxide proxies and ocular perfusion pressure in primary open angle glaucoma. Br. J. Ophthalmol. 2004, 88, 757–760.
- Doganay, S.; Evereklioglu, C.; Turkoz, Y.; Er, H. Decreased nitric oxide production in primary open-angle glaucoma. Eur. J. Ophthalmol. 2002, 12, 44–48.
- Buys, E.S.; Ko, Y.C.; Alt, C.; Hayton, S.R.; Jones, A.; Tainsh, L.T.; Ren, R.; Giani, A.; Clerte, M.; Abernathy, E.; et al. Soluble Guanylate Cyclase alpha1-Deficient Mice: A Novel Murine Model for Primary Open Angle Glaucoma. PLoS ONE 2013, 8, e60156.
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411.
- Pasquale, L.R. Vascular and autonomic dysregulation in primary open-angle glaucoma. Curr. Opin. Ophthalmol. 2016, 27, 94–101.
- Kolluru, G.K.; Tamilarasan, K.P.; Rajkumar, A.S.; Geetha Priya, S.; Rajaram, M.; Saleem, N.K.; Majumder, S.; Ali, B.M.J.; Illavazagan, G.; Chatterjee, S. Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated leakiness. Eur. J. Cell Biol. 2008, 87, 147–161.
- Monica, F.Z.; Bian, K.; Murad, F. The Endothelium-Dependent Nitric Oxide-cGMP Pathway. Adv. Pharmacol. 2016, 77, 1–27.
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp. Neurol. 2004, 190, 446–455.
- Tong, L.; Hill, R.A.; Damisah, E.C.; Murray, K.N.; Yuan, P.; Bordey, A.; Grutzendler, J. Imaging and optogenetic modulation of vascular mural cells in the live brain. Nat. Protoc. 2021, 16, 472–496.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561.
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427.
- Henshall, T.L.; Keller, A.; He, L.; Johansson, B.R.; Wallgard, E.; Raschperger, E.; Mae, M.A.; Jin, S.; Bersholtz, C.; Lendahl, U. Notch3 is necessary for blood vessel integrity in the central nervous system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 409–420.
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783.
- Kutcher, M.E.; Kolyada, A.Y.; Surks, H.K.; Herman, I.M. Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am. J. Pathol. 2007, 171, 693–701.
- Alarcon-Martinez, L.; Shiga, Y.; Villafranca-Baughman, D.; Belforte, N.; Quintero, H.; Di Polo, A. Perciyte dysfunction and loss of inter-pericyte tunneling nanotubes promote neurovascular deficits in glaucoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2110329119.
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95.
- Majumder, S.; Tamilarasan, K.P.; Kolluru, G.K.; Muley, A.; Nair, C.M.; Omanakuttan, A.; Murty, K.V.G.K.; Chatterjee, S. Activated pericyte attenuates endothelial functions: Nitric oxide-cGMP rescues activated pericyte-associated endothelial dysfunctions. Biochem. Cell Biol. 2007, 85, 709–720.
- Newman, E.A. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140195.
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770.
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312.
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sanchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443.
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence Supporting the Existence of an Activity-Dependent Astrocyte-Neuron Lactate Shuttle. Dev. Neurosci. 1998, 20, 291–299.
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43.
- Boal, A.M.; Risner, M.L.; Cooper, M.L.; Wareham, L.K.; Calkins, D.J. Astrocyte Networks as Therapeutic Targets in Glaucomatous Neurodegeneration. Cells 2021, 10, 1368.
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650.
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079.
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896.
- Reichenbach, A.; Bringmann, A. Glia of the human retina. Glia 2020, 68, 768–796.
- Dixon, M.A.; Greferath, U.; Fletcher, E.L.; Jobling, A.I. The Contribution of Microglia to the Development and Maturation of the Visual System. Front. Cell Neurosci. 2021, 15, 659843.
- Varela, H.J.; Hernandez, M.R. Astrocyte responses in human optic nerve head with primary open-angle glaucoma. J. Glaucoma 1997, 6, 303–313.
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in glaucomatous optic neuropathy. Prog. Brain Res. 2008, 173, 353–373.
- Pifarre, P.; Prado, J.; Giralt, M.; Molinero, A.; Hidalgo, J.; Garcia, A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J. Neurochem. 2010, 112, 807–817.
- Bonkale, W.L.; Winblad, B.; Ravid, R.; Cowburn, R.F. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci. Lett 1995, 187, 5–8.
- Baltrons, M.A.; Pifarré, P.; Ferrer, I.; Carot, J.M.; García, A. Reduced expression of NO-sensitive guanylyl cyclase in reactive astrocytes of Alzheimer disease, Creutzfeldt–Jakob disease, and multiple sclerosis brains. Neurobiol. Dis. 2004, 17, 462–472.
- Kusaka, S.; Dabin, I.; Barnstable, C.J.; Puro, D.G. cGMP-mediated effects on the physiology of bovine and human retinal Müller (glial) cells. J. Physiol. 1996, 497 Pt 3, 813–824.
- Paris, D.; Town, T.; Parker, T.A.; Tan, J.; Humphrey, J.; Crawford, F.; Mullan, M. Inhibition of Alzheimer’s β-amyloid induced vasoactivity and proinflammatory response in microglia by a cGMP-dependent mechanism. Exp. Neurol. 1999, 157, 211–221.
- Paris, D.; Town, T.; Mullan, M. Novel strategies for opposing murine microglial activation. Neurosci. Lett. 2000, 278, 5–8.
- Choi, S.H.; Choi, D.H.; Song, K.S.; Shin, K.H.; Chun, B.G. Zaprinast, an inhibitor of cGMP-selective phosphodiesterases, enhances the secretion of TNF-α and IL-1β and the expression of iNOS and MHC class II molecules in rat microglial cells. J. Neurosci. Res. 2002, 67, 411–421.
- Pilz, R.B.; Broderick, K.E. Role of cyclic GMP in gene regulation. Front. Biosci. 2005, 10, 1239–1268.
- Vollmar, A.M. The role of atrial natriuretic peptide in the immune system. Peptides 2005, 26, 1086–1094.
- Moriyama, N.; Taniguchi, M.; Miyano, K.; Miyoshi, M.; Watanabe, T. ANP inhibits LPS-induced stimulation of rat microglial cells by suppressing NF-kappaB and AP-1 activations. Biochem. Biophys Res. Commun. 2006, 350, 322–328.
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 2006, 281, 14971–14980.
- Duan, Y.; Haugabook, S.J.; Sahley, C.L.; Muller, K.J. Methylene blue blocks cGMP production and disrupts directed migration of microglia to nerve lesions in the leech CNS. J. Neurobiol. 2003, 57, 183–192.
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21.
- Borán, M.S.; García, A. The cyclic GMP-protein kinase G pathway regulates cytoskeleton dynamics and motility in astrocytes. J. Neurochem. 2007, 102, 216–230.
- Brahmachari, S.; Fung, Y.K.; Pahan, K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 2006, 26, 4930–4939.
- Holden, J.M.; Al Hussein Al Awamlh, S.; Croteau, L.-P.; Boal, A.M.; Rex, T.S.; Risner, M.L.; Calkins, D.J.; Wareham, L.K. Dysfunctional cGMP Signaling Leads to Age-Related Retinal Vascular Alterations and Astrocyte Remodeling in Mice. Int. J. Mol. Sci. 2022, 23, 3066.
- Malone, P.; Miao, H.; Parker, A.; Juarez, S.; Hernandez, M.R. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia 2007, 55, 1085–1098.
- Ampey, B.C.; Ampey, A.C.; Lopez, G.E.; Bird, I.M.; Magness, R.R. Cyclic Nucleotides Differentially Regulate Cx43 Gap Junction Function in Uterine Artery Endothelial Cells From Pregnant Ewes. Hypertension 2017, 70, 401–411.
- Podda, M.V.; Leone, L.; Piacentini, R.; Cocco, S.; Mezzogori, D.; D’Ascenzo, M.; Grassi, C. Expression of olfactory-type cyclic nucleotide-gated channels in rat cortical astrocytes. Glia 2012, 60, 1391–1405.
- Zarate, S.M.; Huntington, T.E.; Bagher, P.; Srinivasan, R. Aging reduces calreticulin expression and alters spontaneous calcium signals in astrocytic endfeet of the mouse dorsolateral striatum. bioRxiv 2021.
- Woart, N.M. Enhanced cGMP-Dependent Signaling in Astrocytes: Novel Therapeutic Target in Alzheimer’s Disease; Philadelphia College of Osteopathic Medicine: Philadelphia, PA, USA, 2015.
- Jehle, A.; Garaschuk, O. The Interplay between cGMP and Calcium Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7048.
- Burette, A.; Zabel, U.; Weinberg, R.J.; Schmidt, H.H.; Valtschanoff, J.G. Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J. Neurosci. 2002, 22, 8961–8970.
- Wareham, L.K.; Dordea, A.C.; Schleifer, G.; Yao, V.; Batten, A.; Mertz, J.; Gregory-Ksander, M.; Pasquale, L.; Buys, E.S.; Sappington, R.M. Increased bioavailability of cyclic guanylate monophosphate prevents retinal ganglion cell degeneration. Neurobiol. Dis. 2019, 121, 65–75.
- Wareham, L.K.; Buys, E.S.; Sappington, R.M. The nitric oxide-guanylate cyclase pathway and glaucoma. Nitric Oxide 2018, 77, 75–87.
- Fiscus, R.R.; Johlfs, M.G. Protein kinase G (PKG): Involvement in Promoting Neural Cell Survival, Proliferation, Synaptogenesis, and Synaptic Plasticity and the Use of New Ultrasensitive Capillary-Electrophoresis-Based Methodologies for Measuring PKG Expression and Molecular Actions. In Protein Kinase Technologies; Mukai, H., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 319–347.
- Grimes, M.T.; Powell, M.; Gutierrez, S.M.; Darby-King, A.; Harley, C.W.; McLean, J.H. Epac activation initiates associative odor preference memories in the rat pup. Learn. Mem. 2015, 22, 74–82.
- Nagai-Kusuhara, A.; Nakamura, M.; Mukuno, H.; Kanamori, A.; Negi, A.; Seigel, G.M. cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp. Eye Res. 2007, 84, 152–162.
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255.
- Hirooka, K.; Kourennyi, D.E.; Barnes, S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J. Neurophysiol. 2000, 83, 198–206.