2. Clinical Development of Systemic AAV-Mediated Mini/Micro-Dystrophin Gene Therapy
rAAV, derived from the wild-type AAVs, are well characterized as potential gene transfer vehicles in the treatment of neuromuscular disorders, with the capacity to transduce the vast majority of the striated musculature with a single administration. AAV is composed of an icosahedral protein capsid of ~26 nm in diameter and a single-stranded DNA genome of ~4.7 kb that can either be the sense or anti-sense strand. The capsid comprises three types of subunit, VP1, VP2 and VP3, in a ratio of 1:1:10 (VP1:VP2:VP3). The genome is comprised of three genes (
Rep,
Cap and
aap), which are flanked by two T-shaped inverted terminal repeats (ITRs) that largely serve as the viral origins of replication and the packaging signal
[8][25]. The
Rep encodes proteins required for gene replication,
Cap gene encodes the three capsid subunits and recognizes the host cell surface and the
aap gene encodes the assembly-activating protein needed for the capsid assembly and nuclear localization for some AAV serotypes
[9][26]. However, rAAV genomes substitute all the AAV protein-coding sequence with therapeutic gene expression cassettes, and just reserve the ITRs, which are needed for gene replication and packaging during vector production. The complete removal of viral coding sequences maximizes the packaging capacity of rAAVs and contributes to their low immunogenicity and cytotoxicity when delivered in vivo
[10][27].
To circumvent the difficulty in AAV packaging with the full-length dystrophin gene, mini/micro-dystrophin therapies has been developed. The notion of miniaturized dystrophin was based on a Becker muscular dystrophy patient, who remained ambulatory for seven decades despite a deletion of nearly half of the
DMD gene
[11][28]. Recently, mini-dystrophin incorporating different spectrin repeats (SRs) and hinges (
Figure 1) delivered by different AAV serotypes are undergoing assessment of safety and efficacy in three simultaneous gene therapy trials (
Table 1)
[12][29] sponsored by Sarepta Therapeutics, Pfizer, and Solid Biosciences.
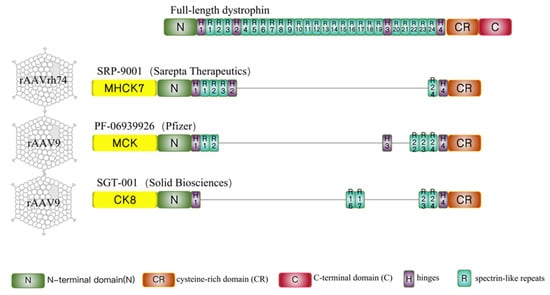
Figure 1. Full-length dystrophin and micro-dystrophins designed in 3 clinical trials. Full-length dystrophin contains an N-terminal domain (N), 24 spectrin-like repeats (R1 to R24), four hinges (H1 to H4), a cysteine-rich domain (CR), and a C-terminal domain (C). SRP-9001(ΔR4-R23/ΔC) is the candidate drug developed by Sarepta Therapeutics and regulated by the MHCK7 promoter. PF-06939926(ΔR3-R21 + H3/ΔC) is used in clinical trials sponsored by Pfizer and driven by the MCK promoter. SGT-001 (ΔR1-R22 + R16R17/ΔC) is a type of micro-dystrophin designed by Solid Biosciences and regulated by the CK8 promoter. The common characteristics of these three micro-dystrophins are the preservation of the N-terminal domain, cysteine-rich domain, and portion of spectrin-like repeats and hinges and the absence of C-terminal domain. The differences are in the central hinges and the R16/17 nNOS-binding domain. SRP-9001 contains hinge 2 and PF-06939926 contains hinge 3, only SGT-001 carries the R16/17 nNOS-binding domain.
Table 1.
Current status of systemic AAV mini/micro-dystrophin clinical trials.
2.1. Sarepta-SRP-9001
Sarepta Therapeutics used rAAVrh74 (a serotype very similar to AAV-8) to delivery of the micro-dystrophin driven by MHCK7 promoter
[13][30], named as SRP-9001. MHCK7 promoter (~770 bp), a novel regulatory cassette based on enhancer/promoter regions of murine muscle creatine kinase (CK) and α-myosin heavy-chain genes, can direct high-level expression comparable to cytomegalovirus and Rous sarcoma virus promoters in fast and slow skeletal and cardiac muscle, and low expression in the liver, lung, and spleen
[13][30]. According to the phase 1/2 open-label, safety, and tolerability trial (ClinicalTrials.gov: NCT03375164), 4 DMD boys, mean age 4.8, with mutations between exons 18 and 58, took prednisolone for ≥12 weeks but the AAVrh74 total binding antibody titers < 1:400 were enrolled
[14][31]. The subjects received a daily dose of 1 mg/kg prednisone for 30 days before the 2.0 × 10
14 vg/kg SRP-9001 was injected. After 12 weeks, therapeutic efficiency was assessed. Compared to the baseline, the gastrocnemius muscle biopsy showed a mean of 81.2% muscle fiber expression of micro-dystrophin, and the mean expression level was 74.3%. Serum CK remained decreased in all subjects (range 46~85%), with functional improvement by a mean 5.5 points in the North Star Ambulatory Assessment (NSAA) at 1 year. Meanwhile, the adverse event (AE) profile was minimal. The transient elevation of γ-glutamyltransferase in three patients was resolved with corticosteroids and the most common was vomiting (50%), which was considered to be not correlated with the AAV immunity. Currently, the treated patients average over 9 years old and in the predicted steep decline phase of disease they did not decline, but demonstrated a 7-point increase above their pre-treatment baselines on NSAA, and a 9.9-point (unadjusted means) and 9.4-point (least squared means) improvement versus a propensity-weighted external control (
p = 0.0125)
[15][32].
NCT03769116 is a randomized double-blind placebo-controlled phase 2 trial assessing the efficacy of SRP-9001 in 41 males with DMD. DMD patients between the ages of 4 and 7 and who have been taking oral corticosteroids for at least 12 weeks prior to the study were enrolled as participants. Participants were treated with a single intravenous injection of either SRP-9001 or a placebo. Primarily, the outcomes analyzed the change in the baseline NSAA score after 48 weeks and the micro-dystrophin expression after 12 weeks. Secondary outcomes assessed the time of the 100 m timed test, time to rise from the floor, and time to ascend four steps, as well as further quantifying micro-dystrophin using IF. After 48 weeks, patients in the treatment group were moved to the placebo group and vice versa, and all patients were treated with intravenous injection once again. The analysis reoccur 48 weeks after this second round of treatment, followed by a 3-year open-label extension for all patients. In January 2021, topline results for the first 48-week period were released
[16][33]. Participants injected with SRP-9001 demonstrated significantly increased micro-dystrophin expression at 12 weeks compared to the baseline (mean expression of 28.1%), and no new safety concerns arose. However, there was no significant improvement of the NSAA score compared to the placebo after 48 weeks. Sarepta suggested that this discrepancy was due to poor randomization, which assigned patients with a higher baseline NSAA to the placebo group, who thus had a better natural history.
In late 2019, Sarepta announced another clinical trial, NCT04626674, known as the ENDEAVOR trial
[17][34]. Thirty-eight patients with the same criteria as in NCT03375164 were confirmed as participants. Interim data from the first 11 patients in this study were released at the World Muscle Society Virtual Congress in September 2021
[18][35]. These data indicated that the SRP-9001 treatment was associated with robust micro-dystrophin expression localized to the sarcolemma and suggest that the level of expression was related to the vector genome copy number. In addition, adverse effects were consistent with previous studies, and all treatment-related adverse effects were temporary and manageable. Shortly after these data were presented, Sarepta released the results of the integrated analysis of the across studies 101, 102, and 103 at the target dose. At one year, SRP-9001 treated patients improved by 3.1 points (unadjusted means) and 2.4 points (least squared means) on NSAA versus the propensity-weighted external control (
p ≤ 0.0001)
[19][36]. Therefore, these provide further support to reinforce confidence in our ongoing Phase 3 Study SRP-9001-301, EMBARK, which is a randomized double-blind placebo-controlled trial designed to assess the efficacy of SRP-9001 treatment for patients with DMD
[20][37].
2.2. Pfizer-PF-06939926
At the American Society for Gene and Cell Therapy (ASGCT) meeting on May 15, 2020, Pfizer’s press released the preliminary results of its ongoing clinical trial (ClinicalTrials.gov: NCT03362502)
[21][38]. This is an open-label study to primarily evaluate the safety and tolerability of PF-06939926. PF-06939926 is a recombinant AAV9 carrying the mini-dystrophin under the control of a minimized murine muscle creatine kinase (MCK) promoter
[22][39]. The preliminary results were collected from nine DMD patients of 4 years and older taking daily glucocorticoids for at least 3 months, with no pre-existing neutralizing antibodies to AAV9 before the injection of PF-06939926. Three patients received a low dose of 1.0 × 10
14 vg/kg, and six patients received a high dose of 3.0 × 10
14 vg/kg. Immunofluorescence (IF) and liquid chromatography mass spectrometry (LCMS) were used to assess the expression of mini-dystrophin. The data indicated that comparing to the normal, 2 months was 20% and 12 months was 24% (
n = 3). At a high dose, the expression was 35% (
n = 6) at 2 months and 52% (
n = 3) at 12 months. Three subjects demonstrated a median improvement of 3.5 points from the baseline in NSAA at 12 months. Fat fraction demonstrated a reduction of 8% at a high dose (
n = 3) and was unchanged at a low dose at 1 year, estimated by MRI. However, more than 40% patients suffered vomiting, nausea, decreased appetite, and pyrexia. Furthermore, three serious adverse events requiring urgent intervention were observed in the first two weeks following treatment, persistent vomiting, resulting in dehydration, acute kidney injury involving atypical hemolytic uremic syndrome (aHUS)-like with complement activation requiring hemodialysis and eculizumab. All three events were effectively treated and had resolved completely within 2 weeks. Then, the trial was originally put on hold by Pfizer to enable protocol amendments.
During a subsequent update at the Muscular Dystrophy Association’s (MDA) Scientific and Clinical Conference in March 2021, Pfizer announced that it had dosed another 10 patients with PF-06939926, for a total of 19
[23][40]. Thankfully, there were no more serious AE presented at this extended cohort, which Pfizer attributed to the implementation of a glucocorticoid-based mitigation plan inspired by its previous interim results. A total of 30% of patients in the larger cohort experienced the previously mentioned minor AE, and improvements in NSAA scores were consistent with the previous findings. Based on these promising findings, PF-06939926 received a fast-track designation from the FDA to begin phase-3 trials in late 2020 (NCT04281485)
[24][41], and the first participant was been dosed in in Barcelona, Spain on December 29, 2020. However, in September 2021, three patients were reported to have experienced serious treatment-related muscle weakness, two of whom also suffered from myocarditis, leading to the exclusion of patients who have mutations in exon 9 through 13 or both exon 29 and 30 from all trials
[25][42]. Unfortunately, in December 2021, Pfizer reported the death of a patient being treated with PF-06939926
[26][43]. No further details have been provided by Pfizer at this point, and both NCT03362502 and the recruitment for the upcoming phase 3 NCT04281485 have been put on hold pending an investigation.
2.3. Solid Biosciences-SGT-001
Solid Biosciences initiated a phase 1/2 open-label clinical trial (ClinicalTrials.gov: NCT03368742) designed to assess the safety and preliminary efficacy in DMD patients treated with SGT-001. SGT-001 was the candidate drug using AAV9 and a CK8 muscle-specific promoter to deliver and drive micro-dystrophin carrying the R16/17 neuronal nitric oxide synthase (nNOS) binding domain, which is considered to allow sufficient blood perfusion in the working muscle
[27][44]. Participants in this study are DMD patients who are males between 4 and 17 years, negative for the AAV9 antibodies, and who have used oral corticosteroids for at least 12 weeks prior to beginning the trial. Interim data from the first six patients were made available via a press release from Solid Biosciences in March 2021. Six subjects were treated by SGT-001 with a low dose (5.0 × 10
13 vg/kg,
n = 3) and high dose (2.0 × 10
14 vg/kg,
n = 3). IF and Western blotting (WB) were used to estimate the expression level of micro-dystrophin. In a patient with the low dose, the micro-dystrophin was below the 5% level of quantification by WB and approximately 10% of fibers by IF. However, in the other two subjects, micro-dystrophin was detected at very minimal levels by IF and none by WB. However, in the three subjects of the high dose, the micro-dystrophin expression ranged from approximately 5–17.5% of the normal dystrophin by WB and 10–70% of positive muscle fibers by IF at day 90. Several days later, the non-ambulatory adolescent DMD patient demonstrated a platelet count reduction, followed by a red blood cell count reduction and transient renal impairment. Patients in the low-dose cohort had a mean NSAA score increase of 1.0, while patients in the high-dose cohort had an increase of 0.3. Patients in the untreated control group experienced a 4-point decline in the same time frame, indicating an initial clinical benefit associated with the treatment with SGT-001. Patients in both dose groups achieved clinical improvements on the 6-min walk test and vital capacity test, as well as meaningful improvements as assessed by patient-reported outcome measures (PROMs). The AEs were serious and the repeated complement activation has resulted in two clinical holds by the FDA. The first was related to thrombocytopenia in 2018 and the second was in October 2019 because of a more widespread complement activation affecting red blood cells (RBCs) and causing renal damage and cardiopulmonary insufficiency
[28][29][45,46]. Due to the small number of patients tested and the difficulties Solid Biosciences faced in this trial, firm conclusions regarding the safety or preliminary efficacy cannot be drawn from this study at this time. An in vitro study suggests that the AAV capsid not only interacted with various components of the complement system, but also directly activated the complement system in a dose-dependent manner
[30][47].