Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Patricia A. J. Muller.
TP53
is mutated in the majority of human cancers. Mutations can lead to loss of p53 expression or expression of mutant versions of the p53 protein. These mutant p53 proteins have oncogenic potential. They can inhibit any remaining WTp53 in a dominant negative manner, or they can acquire new functions that promote tumour growth, invasion, metastasis and chemoresistance.
- mutant p53 chemoresistance
- targeted therapy
- gain-of-function
1. p53 and Mutations in Cancers
p53 is a tumour suppressor protein and nuclear transcription factor (53 kDa) regulating target genes and involved in apoptosis, senescence, cell cycle arrest, and DNA repair. In response to low doses of genomic stress, both extrinsic (e.g., UV-induced DNA damage) and intrinsic (e.g., chromosomal aberrations), p53 regulates cell cycle arrest to allow for DNA repair [1,2,3][1][2][3]. In response to high doses of stress, p53 is more likely to promote apoptosis. Importantly, many chemotherapeutics act by inducing this stress-induced cell death function of p53 to destroy tumour cells.
In the absence of stress, p53 protein expression is kept at low levels [4]. This is facilitated by the E3 ubiquitin ligase MDM-2 (mouse double minute-2) that ubiquitinates p53 leading to its degradation. In response to DNA damage, p53 is released from MDM2 suppression allowing for p53-mediated transcription. MDM-2 limits p53 expression whilst p53 directly promotes MDM-2 expression. This creates an autofeedback loop that allows for a fast and dynamic signalling response to react to differences in stress quickly (Figure 1) [5,6][5][6].

Figure 1. p53 and mouse double minute-2 (MDM2) auto-feedback loop. DNA damage and cellular stress increase p53 expression and facilitate its nuclear import. This allows for p53’s transcriptional activation of target genes, including MDM2. p53-induced MDM2 activation then results in p53 binding to MDM2 and its proteasomal degradation.
TP53 mutation occurs in ~50–60% of all human cancers and can result in both the absence of protein expression or the expression of a mutated protein [7]. p53 mutational status within tumours is heterogeneous and the onset of TP53 mutations can vary greatly in different cancers. As an example, in colorectal [8], breast [9], and pancreatic [10] cancers, TP53 mutation is marked as a late stage tumourigenic event aiding more with tumour progression than with tumour initiation, while in pre-malignant breast lesions [11], hepatocellular carcinoma [12], and in astrocytoma [13] TP53 mutations present during the early stages of tumorigenesis.
Unlike most other tumour suppressor genes, TP53 mutations often affect a single allele with loss of expression from the remaining allele [14]. This occurs via deletion of part of chromosome 17p [15], methylation of the second allele [16], or through additional mutations [17]. Principally, whilst the presence of TP53 mutations span across almost all of its 393 aa residues (Figure 2), the specificity and frequency of the >25,000 registered TP53 mutations can be differential based on the tumour type, with individual mutants often showing different phenotypical changes [18,19][18][19]. Importantly, most mutations are found in the DNA-binding domain (DBD) with six hotspot mutations at codons 175, 245, 248, 249, 273, and 282 (Figure 2) [14,20][14][20].
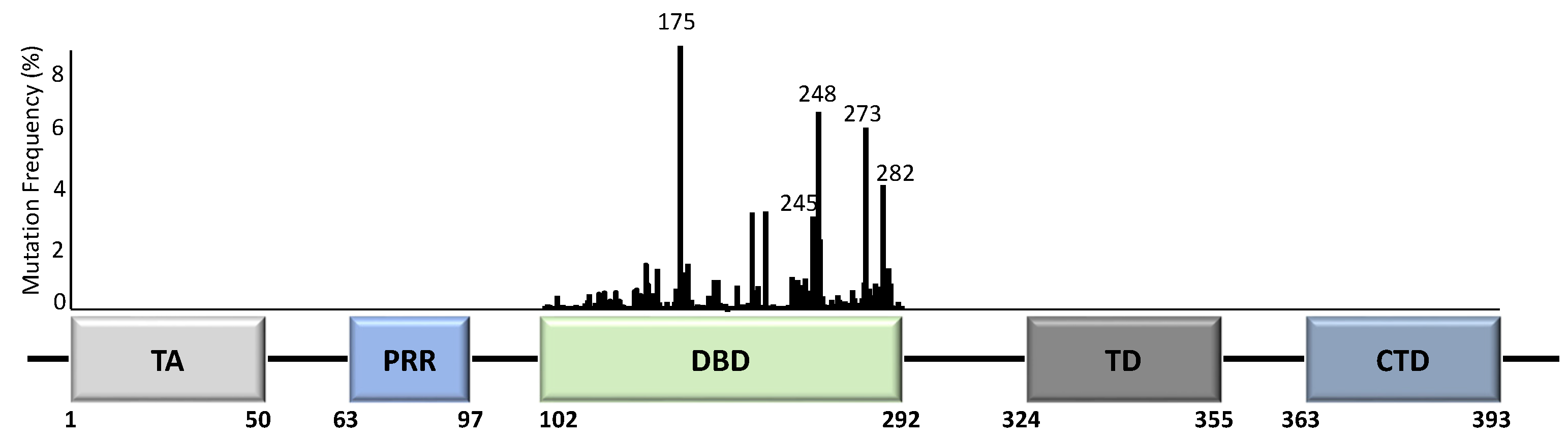
Figure 2. TP53 structure and mutation distribution (%) within the DNA-binding domain (DBD). The frequency of each mutation in all cancers based on the p53 database (www.p53.fr) is indicated for the DBD of TP53. Amino acid positions are indicated below the domains. Five TP53 hotspot mutation sites are further indicated with codon numbers above the bars. TA = transactivation domain; PRR = proline-rich region; DBD = DNA-binding domain; TD = tetramerization domain; CTD = carboxyl terminal regulatory domain.
TP53 mutations can cause truncations or frameshifts in TP53 that almost always result in loss of p53 expression. Missense mutations generally result in expression of mutant proteins with one amino acid variation from WTp53 [14,18][14][18]. This generates a stable mutant p53 protein with longer half-life, seen as increased expression in human cancers [21]. These mutant proteins, including all hotspots, can have alterations in the protein’s structure such as unfolding of the DBD (conformational/structural mutants) [22] or a decreased DNA binding ability (contact mutants) [18].
Mutant p53 proteins often lose some or all of p53’s tumour suppressive function (loss-of-function, LOF) but may also acquire gain-of-function (GOF). This GOF resembles an oncogenic phenotype and is independent of WTp53 [18]. WResearchers and others have shown that mutant p53 promotes invasion and metastasis, tumour growth, genomic instability and chemoresistance [23[23][24][25][26],24,25,26], via a multitude of different mechanisms (reviewed in [27,28,29,30][27][28][29][30]). Mutant p53 proteins can further have dominant-negative effects over the remaining WT protein [20,28][20][28]. This was attributed to mutp53’s ability to form hetero–tetramer complexes with the WTp53 protein [20], causing multimer inactivation [31]. This was seen for both contact and conformational p53 mutants [20].
2. Mutant p53 and Chemoresistance
Mutp53 forms a challenging anti-cancer therapeutic target, mainly due to its lack of druggable allosteric sites, the occurrence of thermodynamically disrupted states as well as its intrinsic ability to confer drug resistance [24,32,33][24][32][33].
The association between mutp53 expression and decreased chemosensitivity is seen in various primary cancers, including breast [34], ovarian [35], lung [36], and hematopoietic [37]. Loss of WTp53 expression can underlie this chemoresistance, but there are also ways in which mutant p53 acquires chemoresistance via its GOF [33]. Mechanisms include, but are not limited to, upregulation of drug transporters, enhanced DNA repair, activation of stemness, apoptosis avoidance, and drug inactivation (Figure 3).
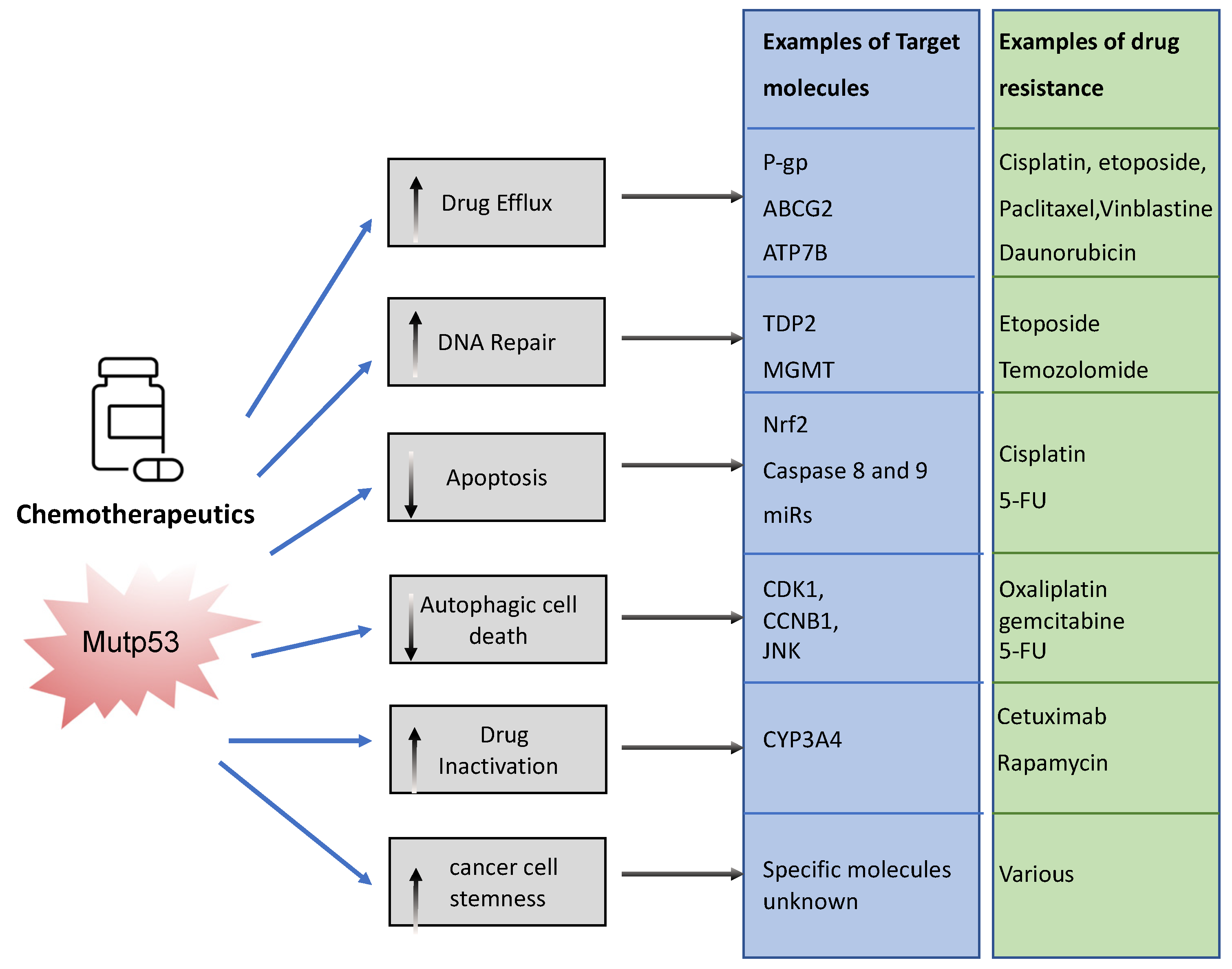
Figure 3. Examples of ways in which mutant p53 promotes chemoresistance. Mutant p53 can impact various cellular processes to prevent chemotherapeutics drugs from working. It can neutralize chemotherapy, promote efflux and impair the way in which these drugs promote cancer cell death. This figure shows examples of these pathways.
To limit toxicity of drugs, mutp53 can directly act on drug availability by regulating drug efflux or drug stability. Mutp53 promotes expression of the (MDR1) gene encoding for the ATP-binding cassette (ABC) transmembrane transporter ABCB1/P-glycoprotein (P-gp) [38]. P-gp extrudes xenobiotic substances/toxic compounds and chemotherapeutic drugs [39]. The transcriptional upregulation of P-gp in cancer is driven by GOF-mutp53’s direct binding to MDR1′s promoter [40]. This enhances drug efflux, reduces drug absorption, and minimizes drug retention/accumulation, causing resistance to anti-cancer drugs, such as taxanes (paclitaxel), vinca alkaloids (vinblastine), and anthracyclines (daunorubicin) [41]. Of note, other ABC transporters such as ABCG2 can also be upregulated by mutant p53 to enhance secretion of 5-flouracil (5-FU) [42]. Interestingly, mutant p53 does not only regulate the gene expression of such transporters. WResearchers recently discovered that mutp53 also specifically enhances plasma membrane expression of P-gp and ATP7B in response to cisplatin and etoposide to enhance efflux, perhaps working in concert with transcriptional regulation of such transporters [24,26][24][26]. GOF-mutp53 (R248W and R282W) can also directly inactivate chemotoxic drugs by upregulating cytochrome P450 enzyme 3A4 (CYP3A4) that help neutralize these drugs [43].
In order to limit toxicity of drugs, mutp53 intercepts in many downstream signalling pathways. In response to drug-induced DNA damage, mutant p53 promotes DNA repair. As an example, GOF-mutp53 (R175H and R248Q) promoted etoposide resistance by enhancing tyrosyl-DNA phosphodiesterase 2′s (TDP2) expression in lung cancer cells in an Ets2 dependent manner [44]. TDP2 in turn repaired etoposide induced double-strand DNA breaks [45], resulting in chemoresistance. Likewise, GOF-mutp53 upregulated the expression of O(6)-methylguanine-DNA-methyltransferase (MGMT) in glioblastoma, enabling the repair of alkylation induced DNA damage by temozolomide [46]. Mutp53 can also directly or indirectly prevent apoptosis. Transcriptionally, it can upregulate Nrf2 (nuclear factor erythroid 2-related factor 2) in response to cisplatin to induce expression of the anti-apoptotic mitochondrial genes: Bcl2 and Bcl-xL [47]. Alternatively, GOF-mutp53’s apoptotic resistance can also occur via direct inhibition of caspases 8 and 9 [48,49,50][48][49][50] or through transcriptional upregulation of miRs that target the apoptosis machinery [51]. Many of the chemotherapeutics are known to cause autophagic cell death through apoptosis. Mutp53 can avoid apoptosis by inducing autophagy via the mTor/AMPK signalling pathway [52], although autophagy itself also regulates mutp53 expression.
It is likely that in an actual cancer, mutant p53 employs one or more of these mechanisms to combat chemotherapeutics, resulting in selection for p53 mutations. In fact, selection of mutp53 is driven by the fact that mutp53 actively promotes stemness [53]. This could be seen as promoting chemoresistance because cancer stem cells are relatively quiescent and therefore less vulnerable to chemotherapy that predominantly acts on highly proliferative cells [53].
References
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528.
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908.
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Methods Mol. Biol. 2013, 962, 49–61.
- Espinosa, J.N.M.; Verdun, R.E.; Emerson, B.M. p53 Functions through Stress- and Promoter-Specific Recruitment of Transcription Initiation Components before and after DNA Damage. Mol. Cell 2003, 12, 1015–1027.
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143.
- Moll, U.M.; Petrenko, O. The MDM2-p53 Interaction. Mol. Cancer Res. 2003, 1, 1001.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767.
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167.
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972.
- Aubele, M.; Werner, M.; Höfler, H. Genetic alterations in presumptive precursor lesions of breast carcinomas. Anal. Cell. Pathol. 2002, 24, 69–76.
- Iakova, P.; Timchenko, L.; Timchenko, N.A. Intracellular signaling and hepatocellular carcinoma. Semin. Cancer Biol. 2011, 21, 28–34.
- Nozaki, M.; Tada, M.; Kobayashi, H.; Zhang, C.L.; Sawamura, Y.; Abe, H.; Ishii, N.; Van Meir, E.G. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neuro. Oncol. 1999, 1, 124–137.
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722.
- Teoh, P.J.; Chung, T.H.; Sebastian, S.; Choo, S.N.; Yan, J.; Ng, S.B.; Fonseca, R.; Chng, W.J. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia 2014, 28, 2066–2074.
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell. Rep. 2019, 28, 1370–1384.e1375.
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249.
- Lu, X. P53: A Target and a Biomarker of Cancer Therapy? Recent Adv. Cancer Res. Ther. 2012, 12, 197–213.
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338.
- Iggo, R.; Gatter, K.; Bartek, J.; Lane, D.; Harris, A.L. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335, 675–679.
- Gannon, J.V.; Greaves, R.; Iggo, R.; Lane, D.P. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. Embo J. 1990, 9, 1595–1602.
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341.
- Phatak, V.; von Grabowiecki, Y.; Janus, J.; Officer, L.; Behan, C.; Aschauer, L.; Pinon, L.; Mackay, H.; Zanivan, S.; Norman, J.C.; et al. Mutant p53 promotes RCP-dependent chemoresistance coinciding with increased delivery of P-glycoprotein to the plasma membrane. Cell Death Dis. 2021, 12, 207.
- Mackay, H.L.; Moore, D.; Hall, C.; Birkbak, N.J.; Jamal-Hanjani, M.; Karim, S.A.; Phatak, V.M.; Pinon, L.; Morton, J.P.; Swanton, C.; et al. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat. Commun. 2018, 9, 3070.
- von Grabowiecki, Y.; Phatak, V.; Aschauer, L.; Muller, P.A.J. Rab11-FIP1/RCP Functions as a Major Signalling Hub in the Oncogenic Roles of Mutant p53 in Cancer. Front. Oncol. 2021, 11, 804107.
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987.
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity: Dominant-negative or gain-of-function? Carcinogenesis 2020, 41, 1635–1647.
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670.
- Malhotra, L.; Singh, A.; Kaur, P.; Ethayathulla, A.S. Comprehensive omics studies of p53 mutants in human cancer. Brief Funct. Genom. 2022, elac015.
- Friedlander, P.; Haupt, Y.; Prives, C.; Oren, M. A mutant p53 that discriminates between p53-responsive genes cannot induce apoptosis. Mol. Cell. Biol. 1996, 16, 4961–4971.
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344.
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant p53 Gain of Function and Chemoresistance: The Role of Mutant p53 in Response to Clinical Chemotherapy. Chemotherapy 2017, 62, 43–53.
- Bergh, J.; Norberg, T.; Sjögren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034.
- Righetti, S.C.; Della Torre, G.; Pilotti, S.; Ménard, S.; Ottone, F.; Colnaghi, M.I.; Pierotti, M.A.; Lavarino, C.; Cornarotti, M.; Oriana, S.; et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996, 56, 689–693.
- Horio, Y.; Takahashi, T.; Kuroishi, T.; Hibi, K.; Suyama, M.; Niimi, T.; Shimokata, K.; Yamakawa, K.; Nakamura, Y.; Ueda, R.; et al. Prognostic significance of p53 mutations and 3p deletions in primary resected non-small cell lung cancer. Cancer Res. 1993, 53, 1–4.
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157.
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462.
- Cascorbi, I. P-glycoprotein: Tissue distribution, substrates, and functional consequences of genetic variations. Handb. Exp. Pharmacol. 2011, 2011, 261–283.
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367.
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127.
- Alam, S.K.; Yadav, V.K.; Bajaj, S.; Datta, A.; Dutta, S.K.; Bhattacharyya, M.; Bhattacharya, S.; Debnath, S.; Roy, S.; Boardman, L.A.; et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016, 23, 707–722.
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108.
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845.
- Ledesma, F.C.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678.
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428.
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705.
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288.
- Ehrhardt, H.; Häcker, S.; Wittmann, S.; Maurer, M.; Borkhardt, A.; Toloczko, A.; Debatin, K.M.; Fulda, S.; Jeremias, I. Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 2008, 27, 783–793.
- Cui, H.; Schroering, A.; Ding, H.F. p53 mediates DNA damaging drug-induced apoptosis through a caspase-9-dependent pathway in SH-SY5Y neuroblastoma cells. Mol. Cancer Ther. 2002, 1, 679–686.
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048.
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061.
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158.
More