Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Jin Ming Zhou.
Covalent inhibitors are a class of small molecule compounds that can covalently bind to specific target proteins, thereby inhibiting their biological functions. Cysteine is one of the least abundant amino acids in proteins of many organisms, which plays a crucial role in catalysis, signal transduction, and redox regulation of gene expression. The thiol group of cysteine possesses the ability to perform nucleophilic and redox-active functions that are not feasible for other natural amino acids. Cysteine is the most common covalent amino acid residue and has been shown to react with a variety of warheads, especially Michael receptors.
- cysteine
- covalent warheads
- covalent inhibitor
1. Introduction
Covalent inhibitors are a class of small molecule compounds that can covalently bind to specific target proteins, thereby inhibiting their biological functions. For a long time, the off-target side effects of covalently bound drugs have been a substantial problem, which limits their potential for drug development [1]. Nonetheless, it is beginning to be recognized that focusing on a specific protein and the mechanism of action, not just the molecular target, but also often the specific binding site and desired mode of action must be specified at the beginning of the project. More and more covalent drugs have been reported and even successfully applied in clinical, such as the proton pump inhibitor omeprazole, the anticoagulant clopidogrel, and the non-small-cell lung cancer inhibitor afatinib, which made people re-recognize the potential of covalent drugs.
Compared with non-covalent inhibitors, one of the most important advantages of covalent inhibitors is their high binding affinity to target proteins, which may result in a relatively long duration of action [2]. In addition, covalent bond inhibitors can also reduce drug resistance caused by target mutations. For example, MRTX849 (adagrasib), developed by Mirati Therapeutics, has been identified as a highly selective covalent inhibitor of KRAS (G12C) and is currently in phase I/II clinical studies. It is an oral selective inhibitor of small molecule KRAS (G12C) mutations. It not only inhibits KRAS mutation almost completely in vivo, but also exhibits favorable drug-like properties [3].
2. Covalent Inhibitors
Recently, various covalent drugs have been emerging. The development of covalent drugs contains many fields such as anticancer, antiviral, and diabetes. In general, covalent binding is much more stable than non-covalent binding. Therefore, compared with classical non-covalent inhibitors, covalent inhibitors have many potential benefits such as prolonging the duration of action, improving ligand efficiency, avoiding drug resistance when targeting amino acids required for enzyme catalysis, and targeting the non-conserved amino acids in high selectivity [4]. Moreover, small-molecule modulators that selectively bind to target proteins can be served not only as tools for understanding protein function but also as clues for drug discovery. While most small molecules function by interacting with their biological targets under equilibrium binding conditions, ligands that chemically modify proteins through the covalent bond formation can offer potential advantages, including improving ligand potency, prolonging the duration of action, enhancing the ability to overcome resistance mutations, and facilitating the identification of target proteins [5]. It has been reported that the covalent reaction-related residues in proteins can be cysteine, lysine [6[6][7],7], glutamic acid [8], serine [9[9][10],10], threonine [11] and tyrosine [12], etc. (Figure 1a). However, the most common covalently reactive residue is the poorly conserved non-catalytic cysteine [13].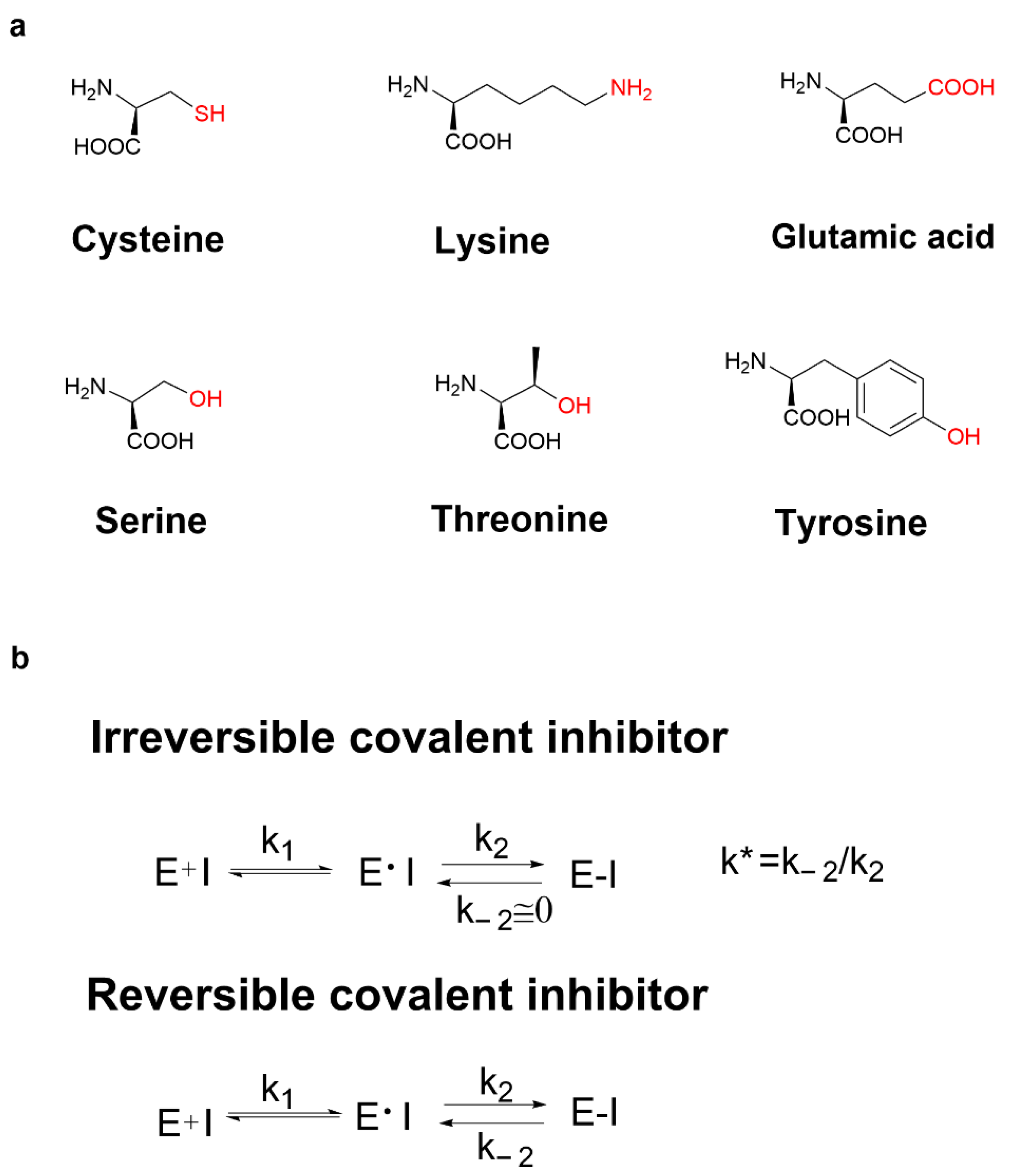
Figure 1. (a) The covalent reaction-related residues; (b) mechanism action of irreversible covalent inhibitors and reversible covalent inhibitors.
3. Covalent Inhibitors Covalently Bound to Cysteine
3.1. Cysteine Profile
Cysteine is one of the least abundant amino acids in proteins of many organisms, which plays a crucial role in the catalysis, signal transduction, and redox regulation of gene expression [14]. Due to the large atomic radius of sulfur and the low dissociation energy of the S-H bond, the thiol group of cysteine possesses the ability to perform nucleophilic and redox-active functions that are not feasible for other natural amino acids. The thioether group in methionine and the sulfhydryl group in neutral amino acids are only moderately nucleophilic; however, once it exists in the cysteine side chain as a thiolate, its nucleophilicity increases by several orders of magnitude, thus becoming the most nucleophilic nucleophile among the 20 common amino acids. Therefore, cysteine has become the most common covalent amino acid residue in covalent drug development [15]. cysteines are very rare, and they have been shown to react with a variety of warheads, especially Michael receptors [16,17][16][17]. These unique properties have led to widespread interest in the nucleophile, leading to the development of a variety of cysteine-targeting warheads with different chemical compositions (Figure 2) [18].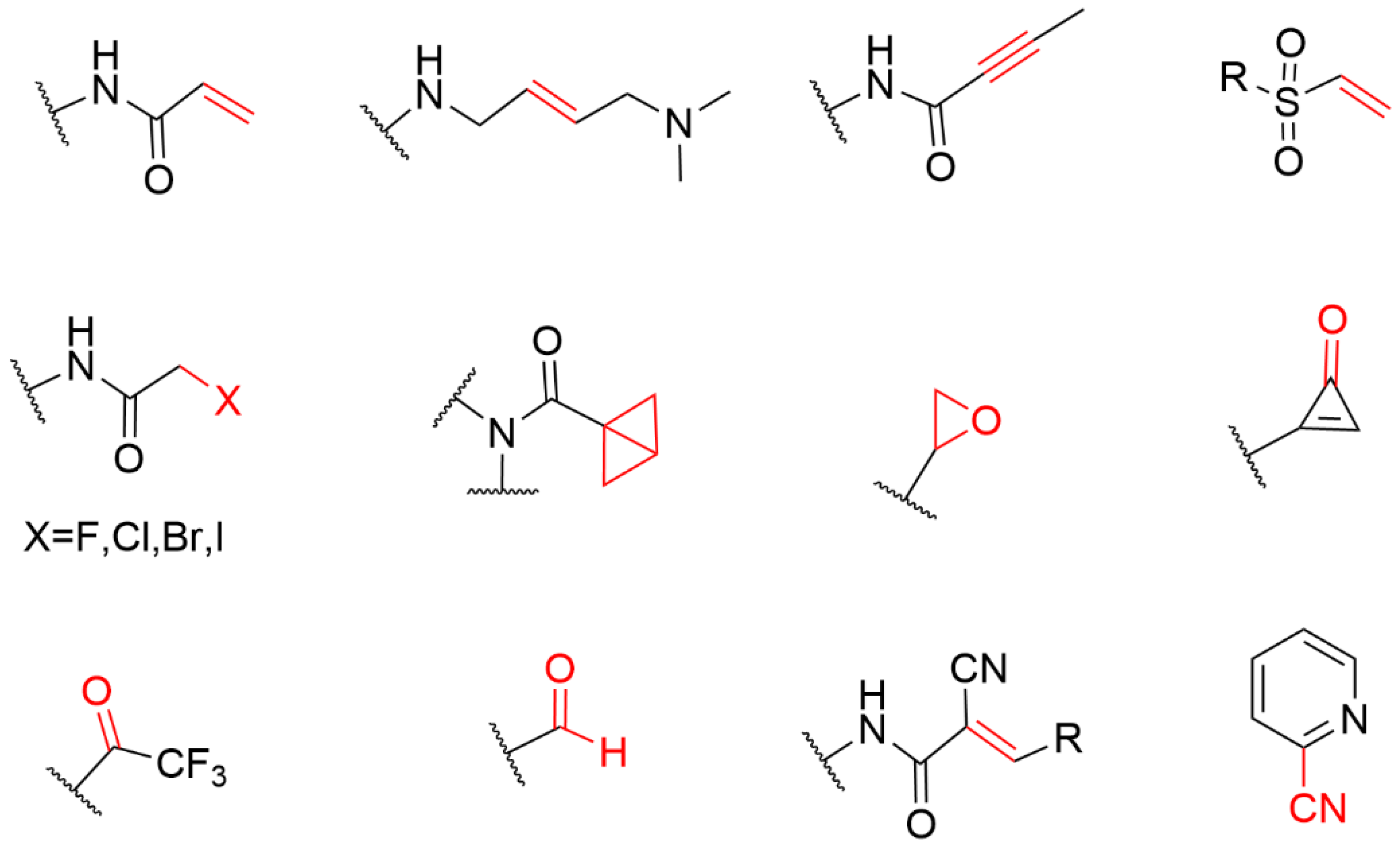
Figure 2.
The general structure of the electrophilic library sorted by warhead chemistries.
3.2. Cysteine-Directed Covalent Drugs
The potency and selectivity of small-molecule drugs can be improved via introducing cysteine-targeting elements that can covalently bind to their targets [13,19][13][19]. Most covalent inhibitors have been designed to target the highly nucleophilic thiol groups of cysteine residues [20]. However, the irreversible nature of these covalent inhibitors might increase the severity of off-target effects, and further optimization of covalent drugs is always required [21]. Recently, there has been mounting interest in developing irreversible inhibitors that can form covalent bonds with cysteines or other nucleophilic residues in the ATP-binding pocket. Gray et al. described the distribution of accessible cysteines and divided cysteines into five groups spanning the entire kinase to facilitate the design of covalent inhibitors. Cysteine residues located in different parts of the binding sites of various targets provided potential opportunities to develop specific irreversible inhibitors using different cysteine positions [22]. Much work in the field of covalent drug development has focused on targeting cysteines with Michael receptors. In particular, acrylamide-based inhibitors have achieved great success. For example, the U.S. Food and Drug Administration (FDA) has approved the marketing of the anticancer drug ibrutinib which covalently inhibited Bruton’s tyrosine kinase (BTK) [23]. Ibrutinib formed irreversible covalent bonds with cysteines near the active site of BTK and has been successfully used to treat B-cell carcinomas [24]. In addition, Gray et al. also reported a selective covalent fibroblast growth factor receptor (FGFR) inhibitor targeting covalent cysteines located at various positions within the ATP-binding pocket, which was of great interest for overcoming the most common resistance to kinase inhibitors [25]. The cyclin-dependent kinase 7 (CDK7) inhibitor THZ1 covalently binds irreversibly to Cys312 of CDK7 located near the kinase domain via the acrylamide moiety [26]. Therefore, both reversible and irreversible inhibitors have the potential to target cysteine. Reversible covalent cysteine-directed drugs have the potential advantage that they are less likely to exhibit off-target effects and instead form covalent adducts with cysteines, thereby increasing specificity and possibly reducing toxicity [27].References
- Uetrecht, J. Idiosyncratic drug reactions: Past, present, and future. Chem. Res. Toxicol. 2008, 21, 84–92.
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531.
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922.
- Jung, Y.; Noda, N.; Takaya, J.; Abo, M.; Toh, K.; Tajiri, K.; Cui, C.; Zhou, L.; Sato, S.-I.; Uesugi, M. Discovery of Non-Cysteine-Targeting Covalent Inhibitors by Activity-Based Proteomic Screening with a Cysteine-Reactive Probe. ACS Chem. Biol. 2022, 17, 340–347.
- Kim, H.-R.; Tagirasa, R.; Yoo, E. Covalent Small Molecule Immunomodulators Targeting the Protease Active Site. J. Med. Chem. 2021, 64, 5291–5322.
- Carrera, A.C.; Alexandrov, K.; Roberts, T.M. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. USA 1993, 90, 442–446.
- Quach, D.; Tang, G.; Anantharajan, J.; Baburajendran, N.; Poulsen, A.; Wee, J.L.K.; Retna, P.; Li, R.; Liu, B.; Tee, D.H.Y.; et al. Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew Chem Int Ed Engl 2021, 60, 17131–17137.
- Martín-Gago, P.; Fansa, E.K.; Winzker, M.; Murarka, S.; Janning, P.; Schultz-Fademrecht, C.; Baumann, M.; Wittinghofer, A.; Waldmann, H. Covalent Protein Labeling at Glutamic Acids. Cell Chem. Biol. 2017, 24, 589–597.
- Powers, J.C.; Asgian, J.L.; Ekici, Ö.D.; James, K.E. Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev. 2002, 102, 4639–4750.
- Fadeyi, O.O.; Hoth, L.R.; Choi, C.; Feng, X.; Gopalsamy, A.; Hett, E.C.; Kyne, R.E.; Robinson, R.P.; Jones, L.H. Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem. Biol. 2017, 12, 2015–2020.
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219.
- Gu, C.; Shannon, D.A.; Colby, T.; Wang, Z.; Shabab, M.; Kumari, S.; Villamor, J.G.; McLaughlin, C.J.; Weerapana, E.; Kaiser, M. Chemical Proteomics with Sulfonyl Fluoride Probes Reveals Selective Labeling of Functional Tyrosines in Glutathione Transferases. Chem. Biol. 2013, 20, 541–548.
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159.
- Pace, N.J.; Weerapana, E. Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol. 2013, 8, 283–296.
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724.
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114.
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α, β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885.
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107.
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073.
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.-M.; Winssinger, N. Cysteine Mapping in Conformationally Distinct Kinase Nucleotide Binding Sites: Application to the Design of Selective Covalent Inhibitors. J. Med. Chem. 2011, 54, 1347–1355.
- Visscher, M.; Arkin, M.R.; Dansen, T.B. Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol. 2016, 30, 61–67.
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889.
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116.
- Tucker, D.L.; Rule, S.A. A critical appraisal of ibrutinib in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. Ther. Clin. Risk Manag. 2015, 11, 979–990.
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877.
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620.
- Johansson, M.H. Reversible michael additions: Covalent inhibitors and prodrugs. Mini-Rev. Med. Chem. 2012, 12, 1330–1344.
More