Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Qinghuai Liu.
Diabetic retinopathy (DR) is currently one of the common causes of vision loss in working-age adults. It is clinically diagnosed and classified according to the vascular changes in the fundus. The activation of immune cells occurs before these vascular changes become detectable. These, together with molecular studies and the positive clinical outcomes of anti-inflammatory treatment, highlight the pivotal involvement of the immune system. The role of innate immunity in DR pathophysiology has been studied in depth, but the contribution of adaptive immunity remains largely elusive.
- adaptive immunity
- diabetic retinopathy
- lymphocyte
1. Introduction
Diabetic retinopathy (DR) is the most common chronic microvascular complication of uncontrolled diabetes and can lead to preventable blindness. Clinically, DR is currently divided into two stages: non-proliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR). NPDR represents the early stages of DR, and its pathological manifestation involves the loss of pericytes, the formation of acellular capillaries and the thickening of basement membranes [1]. Its molecular mechanism is characterized by the activation of endothelial cells; leukocyte stasis, which further leads to endothelial cell dysfunction; and the decomposition of the blood–retinal barrier (BRB). PDR is the more advanced stage of DR and is characterized by the formation of new blood vessels, resulting in retinal hemorrhage and, eventually, irreversible blindness.
2. Adaptive Immunity and DR
2.1. Changes in Adaptive Immunity during DR: Clinical and Experimental Evidence
Signs of lymphocyte activation were observed in circulation in DR patients, including the reduced surface expression of L-selectin. The reduced expression of L-selectin on CD3+ cells was associated with increased leucocyte adhesion. More CD4+ Tfh cells were found in circulation in DR patients [16,17][2][3] and in the lymph nodes and retinal tissues of STZ-induced diabetic mice. The inhibition of Bcl-6, a key transcription factor for Tfh cell development, prevented the up-regulation of Tfh cells and their typical IL-21 cytokines and improved vascular leakage in DR mice or retinal angiogenesis in oxygen-induced retinopathy (OIR) mice [18][4]. These results suggest the involvement of lymphocyte activation in DR pathogenesis. In addition, lymphocyte infiltration was also found in the peripheral samples of DR patients. The densities of CD4+ T cells, CD8+ T cells and CD19+ B cells significantly increased in the FVMs of active PDR patients compared with that of epiretinal membranes without DR [19,20][5][6]. Marked alterations in the lymphocytes of DR patients indicate the strong correlation of lymphocytes with the occurrence and progression of DR. Therefore, how lymphocytes activate and function becomes questions in the puzzle called DR pathophysiology.2.2. Mechanism of Lymphocyte Activation in Diabetes
Two separate signals are required in the complete activation of lymphocytes. Preliminary antigen-specific signals are sent through antigen receptors: the T-cell receptor (TCR) on T cells and the surface immune globulin (Ig) on B cells. The second signal, called co-stimulation, is independent of antigen receptors and is required for full activation. Well-known costimulatory molecules include B7-1 (CD80) and B7-2 (CD86), expressed on activated antigen-presenting cells (APC). Their ligand, CD28, is correspondently found on the surface of T cells [21][7]. Obesity is the hallmark of metabolic syndrome and predisposes patients to the development of major chronic metabolic diseases including type 2 diabetes mellitus (T2DM). In adipose tissue, phenotypic changes in T cells, as well as the recruitment of B and T cells, preceded the infiltration of macrophages [22][8]. A study demonstrated that patients with diabetic ketoacidosis (DKA) and hyperglycemia exhibited increased levels of proinflammatory cytokines and activated CD4+ and CD8+ T lymphocytes [23][9]. In in vitro studies, the expression of CD28-like costimulatory molecule ICOS (CD278) on the surface of T cells was induced by high glucose (HG) levels or advanced glycation end products (AGEs). Higher levels of IFN-γ, interleukin-4 (IL-4), IL-10 and other cytokines were found in the supernatant when T cells were co-cultured with ICOSL-expressing human umbilical vein endothelial cells (HUVECs) but were not observed in non-contact T cells or T cells treated with HUVEC conditioned medium [24][10]. This indicates that direct cell–cell contact between T lymphocytes and endothelial cells is necessary for T cell activation in HG or AGE treatment. Evidence that elevated major histocompatibility complex class II (MHC II) in insulin-resistant tissues is involved in the activation of CD4+ T cells is mounting. A study by Shirakawa et al. demonstrated that activated CD4+ T cells increased in the visceral adipose tissue of obese mice [25][11]. These cells, called adipocyte resident T cells (ART), regulated the metabolic and inflammatory response, mainly through the up-regulation of MHC II and the secretion of leptin by adipocytes to activate CD4+ T cells and subsequently triggered adipose tissue inflammation. MHCII−/− mice fed a high fat diet (HFD) had less adipose inflammation and insulin resistance than wild-type mice [26][12]. These studies suggest that CD4+T, which plays an important role in inflammation and insulin resistance, can be activated by adipocytes in an antigen-specific, contact-dependent direct pathway. In the diabetic state, low levels of effective insulin concentrations as well as high contents of glucose and free fatty acids provide an environment for oxidative stress and inflammatory pathway activation [27][13]. High levels of glucose can form intracellular oxidants in lymphocytes, activating kinase phosphorylates such as IκB, which release nuclear factor-kappaB (NF-κB) into the nucleus. NF-κB binds to κB binding sites in the promoter region of inflammatory genes, leading to the transcription and translation of several proinflammatory mediators [28][14]. Proinflammatory mediators, such as cytokines, adhesion molecules (intercellular adhesion molecule (ICAM) and platelet endothelial cell adhesion molecule (PECAM)), prostaglandins and arachidonic acid, subsequently lead to the activation of T lymphocytes. In conclusion, the activation of lymphocytes in DR is mainly involved in two pathways, including a direct contact pathway (up-regulated MHC II molecules or co-stimulators) and an indirect paracrine pathway (a higher level of cytokines), in a diabetic environment (Figure 1).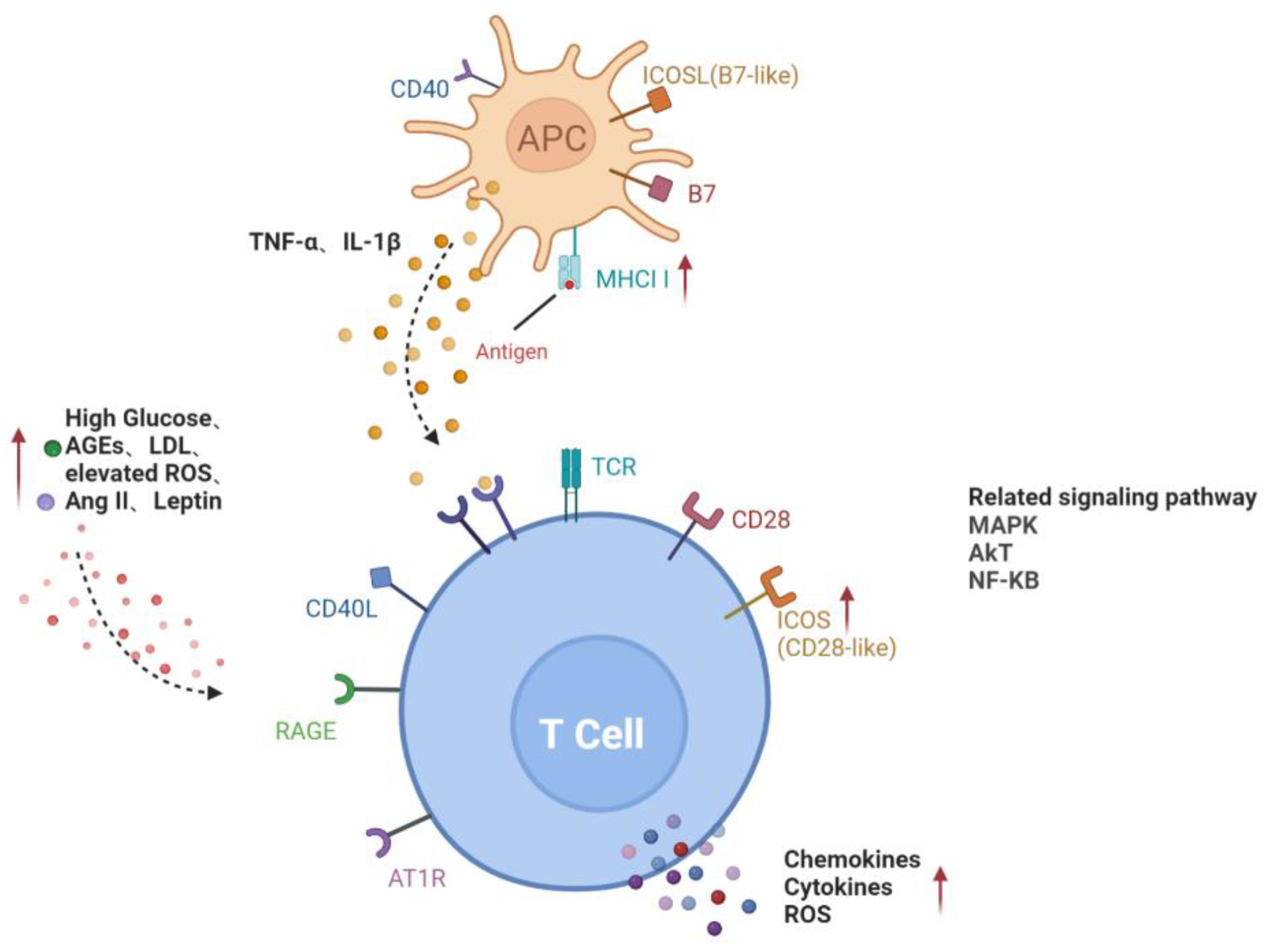
Figure 1. Activation pathways of T lymphocytes in diabetes. Direct contact between APC and T cells through MHCII and co-stimulators on the cell surface; increased glucose, AGEs, LDL, ROS, Ang II and leptin in diabetic microenvironment or cytokines such as TNF-α and IL-1β secreted by APC activate T cells, forming the paracrine pathway. Red arrows represent the upregulation of gene expression or increase of risk factors in diabetes. Black dotted arrows stand for cellular interaction through paracrine pathway. AGEs, advanced glycation end-products; AT1R, angiotensin II type 1 receptor; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; ICOSL, inducible co-stimulatory molecule ligand; MHCII, major histocompatibility complex class II; MAPK, mitogen-activated protein kinase; AKT, v-akt murine thymoma viral oncogene homologue; NF-KB, nuclear factor-kappaB.
2.3. Interaction between Lymphocytes and Retinal Vascular Unit in DR
Retinal vascular dysfunction can be caused by a variety of factors, such as AGE products and receptors, proinflammatory cytokines and chemokines, proliferator-activated receptor-γ disruption, growth factors, oxidative stress and microRNA [29,30,31,32,33,34,35][15][16][17][18][19][20][21]. These factors promote retinal endothelial apoptosis and neovascularization, leading to the development of DR. Lymphocyte infiltration also contributes to the formation of prooxidative and proinflammatory microenvironments, which cannot be neglected in the progression of diabetic microangiopathy. Lymphocytes affect target cells by secreting inflammatory cytokines. Once activated, Th1 cells release large amounts of cytokines such as IFN-γ, IL-2 and TNF, thereby triggering cell-mediated immune response and phagocyte-dependent inflammation [18][4]. Increased levels of IFN-γ and IL-2 have been observed in the vitreous or aqueous humor of patients with diabetes or DR [36,37][22][23] and in retina of diabetic rats [38][24]. Endothelial barrier function is tightly controlled by a wide range of signaling cascades, including the nitric oxide (NO)/cyclic guanosine monophosphate (GMP) pathway. IFN-γ was also demonstrated to promote the permeability of HUVECs, which may be associated with an increase in NO production by IFN-γ [39][25]. In addition, IFN-γ induced disruptions in the endothelial cell junction and reduced the endothelial barrier properties via Rho kinase (ROCK)-mediated cytoskeleton contraction. The neutralization of IFN-γ attenuated vascular ROCK activity and, therefore, improved the endothelial barrier integrity and pericyte coverage [40][26]. The level of TNF-α significantly increased in DR patients, as shown in a meta-analysis study [41][27]. TNF-α reduced the expression of tight junction proteins by activating protein kinase C zeta (PKCζ) and NF-κB, thereby increasing the permeability of retinal endothelial cells [42][28]. In addition, the proinflammatory cytokine IL-17A, mainly produced by Th17 cells, may participate in the development of retinal inflammation and long-term vascular pathology in diabetic mice. T cells and neutrophils expressing IL-17A were found in diabetic retinal vasculature. Furthermore, the IL-17A receptor was expressed on Müller glia cells, retinal endothelial cells and photoreceptors [43][29]. IL-17A signaling induced photoreceptor cells to produce ROS, and systemic ablation of IL-17A reduced retinal inflammation, oxidative stress and vascular leakage. In addition, IL-17A ablation was related to significant reductions in the levels of chemokines (CCL2, CCL5, CX3CL1, CXCL1 and CXCL5), proinflammatory cytokines (TNF and IL-1) and growth factors (G-CSF and GM-CSF) and to increases in the levels of the matrix metalloproteinase 9 (MMP-9) inhibitor and the tissue inhibitor of metalloproteinase (TIMP-1) [43][29]. Among them, MMP-9 is related to the release of TGF-β1 and VEGF and can participate in the destruction of the blood–retina barrier (BRB), inflammation, angiogenesis, mitochondrial damage and apoptosis by changing the vascular permeability [44][30] (Figure 2).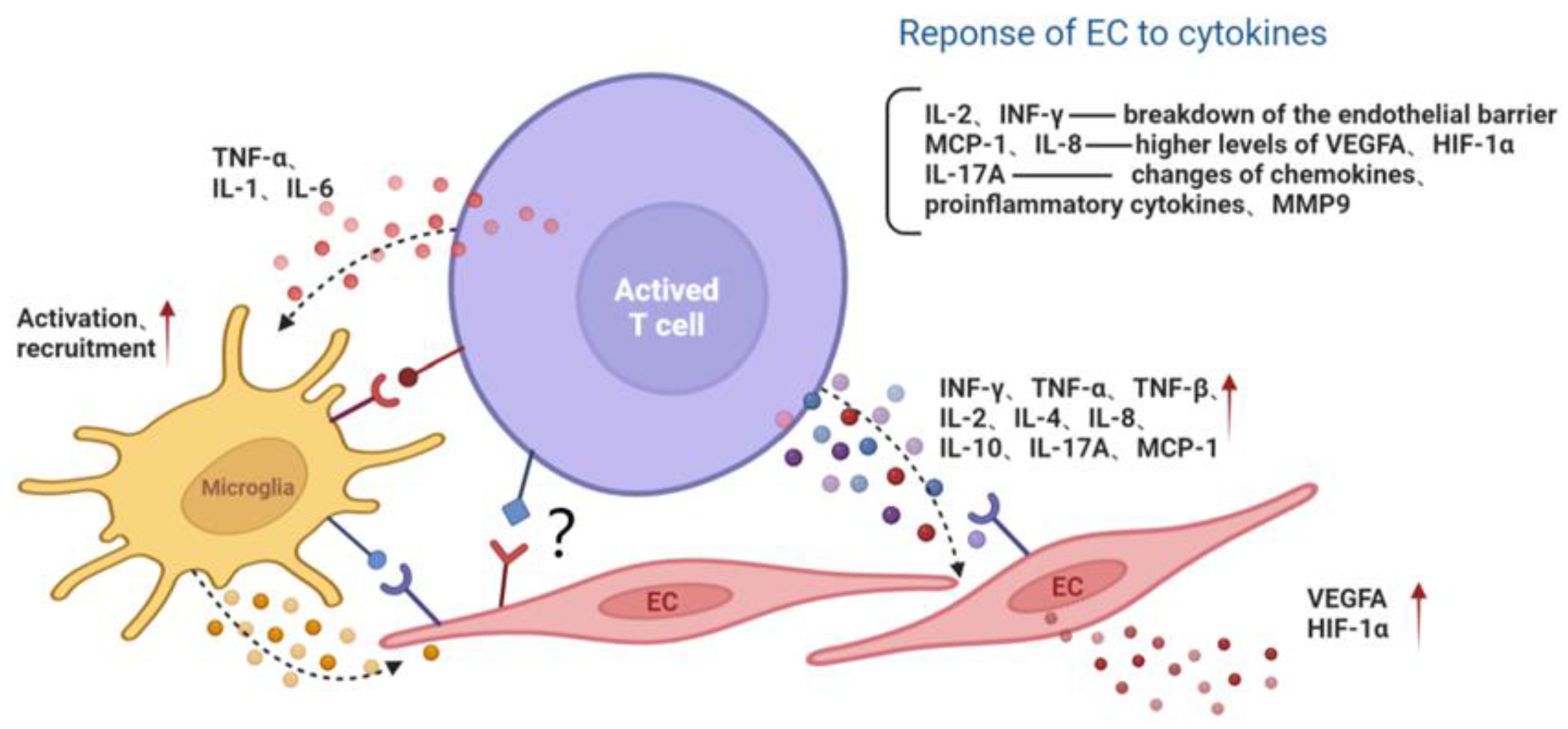
Figure 2. The interaction among activated T lymphocyte, endothelial cell and microglia. Activated T cells secrete cytokines or directly contact microglia and the EC, aggravating the inflammatory microenvironment and endothelial dysfunction. Red arrows represent the upregulation of gene expression. Black dotted arrows stand for cellular interaction through paracrine pathway. EC, endothelial cell; MMP9, matrix metalloproteinase 9; MCP-1, monocyte chemoattractant protein-1; INF, interferon; TNF, tumor necrosis factor; IL, interleukin; VEGFA, vascular endothelial growth factor A; HIF-1α, hypoxia inducible factor-1α.
References
- Pitale, P.M.; Gorbatyuk, M.S. Diabetic Retinopathy: From Animal Models to Cellular Signaling. Int. J. Mol. Sci. 2022, 23, 1487.
- Heuts, F.; Edner, N.M.; Walker LSFollicular, T. Helper Cells: A New Marker of Type 1 Diabetes Risk? Diabetes 2017, 66, 258–260.
- Kenefeck, R.; Wang, C.J.; Kapadi, T.; Wardzinski, L.; Attridge, K.; Clough, L.E.; Heuts, F.; Kogimtzis, A.; Patel, S.; Rosenthal, M.; et al. Follicular helper T cell signature in type 1 diabetes. J. Clin. Investig. 2015, 125, 292–303.
- Liu, Y.; Yang, Z.; Lai, P.; Huang, Z.; Sun, X.; Zhou, T.; He, C.; Liu, X. Bcl-6-directed follicular helper T cells promote vascular inflammatory injury in diabetic retinopathy. Theranostics 2020, 10, 4250–4264.
- Urbančič, M.; Štunf, Š.; Milutinović Živin, A.; Petrovič, D.; GlobočnikPetrovič, M. Epiretinal membrane inflammatory cell density might reflect the activity of proliferative diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8576–8582.
- Urbančič, M.; Petrovič, D.; Živin, A.M.; Korošec, P.; Fležar, M.; Petrovič, M.G. Correlations between vitreous cytokine levels and inflammatory cells in fibrovascular membranes of patients with proliferative diabetic retinopathy. Mol. Vis. 2020, 26, 472–482.
- Azuma, M.; Phillips, J.H.; Lanier, L.L. CD28- T lymphocytes. Antigenic and functional properties. J. Immunol. 1993, 150, 1147–1159.
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716.
- Stentz, F.B.; Kitabchi, A.E. Hyperglycemia-induced activation of human T-lymphocytes with de novo emergence of insulin receptors and generation of reactive oxygen species. Biochem. Biophys. Res. Commun. 2005, 335, 491–495.
- Zhang, H.Y.; Ruan, L.B.; Li, Y.; Yang, T.R.; Liu, W.J.; Jiang, Y.X.; Li, T.R.; Quan, J.; Xuan, W. ICOS/ICOSL upregulation mediates inflammatory response and endothelial dysfunction in type 2 diabetes mellitus. Eur. Rev. Med. Pharm. Sci. 2018, 22, 8898–8908.
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 2016, 126, 4626–4639.
- Deng, T.; Lyon, C.J.; Minze, L.J.; Lin, J.; Zou, J.; Liu, J.Z.; Ren, Y.; Yin, Z.; Hamilton, D.J.; Reardon, P.R.; et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 2013, 17, 411–422.
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820.
- Huang, W.; Xu, L.; Zhou, X.; Gao, C.; Yang, M.; Chen, G.; Zhu, J.; Jiang, L.; Gan, H.; Gou, F.; et al. High glucose induces activation of NF-κB inflammatory signaling through IκBα sumoylation in rat mesangial cells. Biochem. Biophys. Res. Commun. 2013, 438, 568–574.
- Yuuki, T.; Kanda, T.; Kimura, Y.; Kotajima, N.; Tamura, J.; Kobayashi, I.; Kishi, S. Inflammatory cytokines in vitreous fluid and serum of patients with diabetic vitreoretinopathy. J. Diabetes Complicat. 2001, 15, 257–259.
- Miyamoto, K.; Hiroshiba, N.; Tsujikawa, A.; Ogura, Y. In vivo demonstration of increased leukocyte entrapment in retinal microcirculation of diabetic rats. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2190–2194.
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942.
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 2017, 187, 9–19.
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239.
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxidative Med. Cell Longev. 2017, 2017, 9208489.
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.Z. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010, 59, 2297–2305.
- Vujosevic, S.; Micera, A.; Bini, S.; Berton, M.; Esposito, G.; Midena, E. Proteome analysis of retinal glia cells-related inflammatory cytokines in the aqueous humour of diabetic patients. Acta Ophthalmol. 2016, 94, 56–64.
- Tsai, T.; Kuehn, S.; Tsiampalis, N.; Vu, M.K.; Kakkassery, V.; Stute, G.; Dick, H.B.; Joachim, S.C. Anti-inflammatory cytokine and angiogenic factors levels in vitreous samples of diabetic retinopathy patients. PLoS ONE 2018, 13, e0194603.
- Johnsen-Soriano, S.; Sancho-Tello, M.; Arnal, E.; Navea, A.; Cervera, E.; Bosch-Morell, F.; Miranda, M.; Javier Romero, F. IL-2 and IFN-gamma in the retina of diabetic rats. Graefes. Arch. Clin. Exp. Ophthalmol. 2010, 248, 985–990.
- Ng, C.T.; Fong, L.Y.; Low, Y.Y.; Ban, J.; Hakim, M.N.; Ahmad, Z. Nitric oxide participates in IFN-gamma-induced HUVECs hyperpermeability. Physiol. Res. 2016, 65, 1053–1058.
- Bonney, S.; Seitz, S.; Ryan, C.A.; Jones, K.L.; Clarke, P.; Tyler, K.L.; Siegenthaler, J.A. Gamma Interferon Alters Junctional Integrity via Rho Kinase, Resulting in Blood-Brain Barrier Leakage in Experimental Viral Encephalitis. mBio 2019, 10, e01675-19.
- Gao, W.; Zhu, R.; Yang, L. Association of Tumor Necrosis Factor-Alpha-308 G/A and -238 G/A Polymorphism with Diabetic Retinopathy: A Systematic Review and Updated Meta-Analysis. Ophthalmic. Res. 2021, 64, 903–915.
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrósio, A.F.; Antonetti, D.A. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882.
- Sigurdardottir, S.; Zapadka, T.E.; Lindstrom, S.I.; Liu, H.; Taylor, B.E.; Lee, C.A.; Kern, T.S.; Taylor, P.R. Diabetes-mediated IL-17A enhances retinal inflammation, oxidative stress, and vascular permeability. Cell Immunol. 2019, 341, 103921.
- Kowluru, R.A.; Mishra, M. Regulation of Matrix Metalloproteinase in the Pathogenesis of Diabetic Retinopathy. Prog. Mol. Biol. Transl. Sci. 2017, 148, 67–85.
More