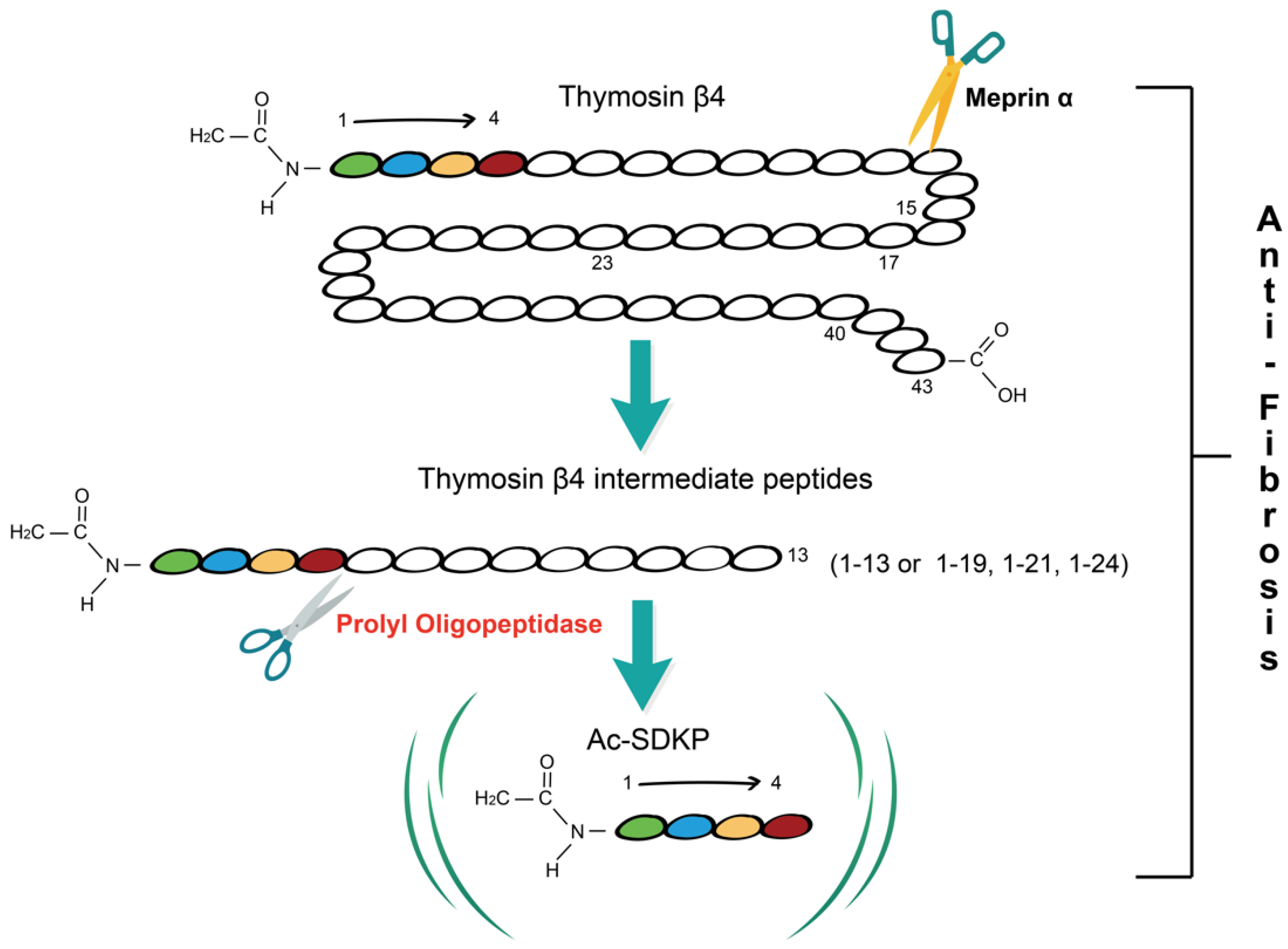
Fibrosis is a pathological process in which parenchymal cells are necrotic and excess extracellular matrix (ECM) is accumulated due to dysregulation of tissue injury repair. Thymosin β4 (Tβ4) is a 43 amino acid multifunctional polypeptide that is involved in wound healing. Prolyl oligopeptidase (POP) is the main enzyme that hydrolyzes Tβ4 to produce its derivative N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) which is found to play a role in the regulation of fibrosis. Accumulating evidence suggests that the Tβ4-POP-Ac-SDKP axis widely exists in various tissues and organs including the liver, kidney, heart, and lung, and participates in the process of fibrogenesis. The Tβ4-POP-Ac-SDKP axis exerts protective effects against organ fibrosis. It is promising that appropriate dosing regimens that rely on this axis could serve as a new therapeutic strategy for alleviating organ fibrosis in the early and late stages.
- Tβ4
- POP
- Ac-SDKP
- fibrosis
1. Tβ4-POP-Ac-SDKP Axis
2. Tissue and Organ Fibrosis
References
- Low, T.L.; Hu, S.K.; Goldstein, A.L. Complete amino acid sequence of bovine thymosin beta 4: A thymic hormone that induces terminal deoxynucleotidyl transferase activity in thymocyte populations. Proc. Natl. Acad. Sci. USA 1981, 78, 1162–1166.
- Xing, Y.; Ye, Y.; Zuo, H.; Li, Y. Progress on the Function and Application of Thymosin β4. Front. Endocrinol. 2021, 12, 767785.
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182.
- Morris, D.; Chopp, M.; Zhang, L.; Lu, M.; Zhang, Z. Thymosin β4 improves functional neurological outcome in a rat model of embolic stroke. Neuroscience 2010, 169, 674–682.
- Shah, R.; Reyes-Gordillo, K.; Cheng, Y.; Varatharajalu, R.; Ibrahim, J.; Lakshman, M.R. Thymosin β4 Prevents Oxidative Stress, Inflammation, and Fibrosis in Ethanol- and LPS-Induced Liver Injury in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 9630175.
- Goldstein, A.L.; Hannappel, E.; Sosne, G.; Kleinman, H.K. Thymosin β4: A multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin. Biol. Ther. 2011, 12, 37–51.
- Ehrlich, H.P.; Iii, S.W.H. Thymosin β4 enhances repair by organizing connective tissue and preventing the appearance of myofibroblasts. Ann. N. Y. Acad. Sci. 2010, 1194, 118–124.
- Cavasin, M.A.; Rhaleb, N.-E.; Yang, X.-P.; Carretero, O.A. Prolyl Oligopeptidase Is Involved in Release of the Antifibrotic Peptide Ac-SDKP. Hypertension 2004, 43, 1140–1145.
- Kumar, N.; Nakagawa, P.; Janic, B.; Romero, C.; Worou, M.E.; Monu, S.R.; Peterson, E.L.; Shaw, J.; Valeriote, F.; Ongeri, E.M.; et al. The anti-inflammatory peptide Ac-SDKP is released from thymosin-β4 by renal meprin-α and prolyl oligopeptidase. Am. J. Physiol.-Renal Physiol. 2016, 310, F1026–F1034.
- Chen, Y.-W.; Liu, B.-W.; Zhang, Y.-J.; Chen, Y.-W.; Dong, G.-F.; Ding, X.-D.; Xu, L.-M.; Pat, B.; Fan, J.-G.; Li, D.-G. Preservation of basal AcSDKP attenuates carbon tetrachloride-induced fibrosis in the rat liver. J. Hepatol. 2010, 53, 528–536.
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and Mechanism of Action of Haloalkanes: Carbon Tetrachloride as a Toxicological Model. Crit. Rev. Toxicol. 2003, 33, 105–136.
- Cavasin, M.A.; Liao, T.-D.; Yang, X.-P.; Yang, J.J.; Carretero, O.A. Decreased Endogenous Levels of Ac-SDKP Promote Organ Fibrosis. Hypertension 2007, 50, 130–136.
- Azizi, M.; Rousseau, A.; Ezan, E.; Guyene, T.T.; Michelet, S.; Grognet, J.M.; Lenfant, M.; Corvol, P.; Ménard, J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J. Clin. Investig. 1996, 97, 839–844.
- Liu, J.-M.; Lawrence, F.; Kovacevic, M.; Bignon, J.; Papadimitriou, E.; Lallemand, J.-Y.; Katsoris, P.; Potier, P.; Fromes, Y.; Wdzieczak-Bakala, J. The tetrapeptide AcSDKP, an inhibitor of primitive hematopoietic cell proliferation, induces angiogenesis in vitro and in vivo. Blood 2003, 101, 3014–3020.
- Lenfant, M.; Wdzieczak-Bakala, J.; Guittet, E.; Prome, J.C.; Sotty, D.; Frindel, E. Inhibitor of hematopoietic pluripotent stem cell proliferation: Purification and determination of its structure. Proc. Natl. Acad. Sci. USA 1989, 86, 779–782.
- Zhang, L.; Xu, L.-M.; Chen, Y.-W.; Ni, Q.-W.; Zhou, M.; Qu, C.-Y.; Zhang, Y. Antifibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline on bile duct ligation induced liver fibrosis in rats. World J. Gastroenterol. 2012, 18, 5283–5288.
- Peng, H.; Carretero, O.A.; Peterson, E.L.; Rhaleb, N.-E. Ac-SDKP inhibits transforming growth factor-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1357–H1364.
- Xu, H.; Yang, F.; Sun, Y.; Yuan, Y.; Cheng, H.; Wei, Z.; Li, S.; Cheng, T.; Brann, D.; Wang, R. A New Antifibrotic Target of Ac-SDKP: Inhibition of Myofibroblast Differentiation in Rat Lung with Silicosis. PLoS ONE 2012, 7, e40301.
- Omata, M.; Taniguchi, H.; Koya, D.; Kanasaki, K.; Sho, R.; Kato, Y.; Kojima, R.; Haneda, M.; Inomata, N. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline Ameliorates the Progression of Renal Dysfunction and Fibrosis in WKY Rats with Established Anti–Glomerular Basement Membrane Nephritis. J. Am. Soc. Nephrol. 2006, 17, 674–685.
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402.
- di Carlo, S.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63.
- Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594.
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321.
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210.
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566.
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1049–1060.
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76.
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93.
- Gur, C.; Wang, S.Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 2022, 185, 1373–1388.e1320.
- Ruaro, B.; Soldano, S.; Smith, V.; Paolino, S.; Contini, P.; Montagna, P.; Pizzorni, C.; Casabella, A.; Tardito, S.; Sulli, A.; et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376.
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873.
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2018, 65, 2–15.