Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Houria Daimi and Version 3 by Diego Franco.
Atrial fibrillation (AF) is known to be the most common supraventricular arrhythmia affecting up to 1% of the general population. Its prevalence exponentially increases with age and could reach up to 8% in the elderly population. The management of AF is a complex issue that is addressed by extensive ongoing basic and clinical research. AF centers around different types of disturbances, including ion channel dysfunction, Ca2+-handling abnormalities, and structural remodeling.
- atrial fibrillation
- gene regulation
- epigenetics
1. microRNAs and Atrial Fibrillation
MicroRNAs (miRNAs) are small (~19–25 nt) non-coding RNAs that are encoded by nuclear DNA and transcribed by RNA polymerase II. miRNAs main function is regulating gene expression post-transcriptionally through binding to complementary target sites within mRNAs, normally within the 3′UTR. This generally results in the inhibition of translation and/or degradation of the target transcript [1][2][135,136]. At present, multiple microRNAs have been involved in electrical and structural remodeling directly linked to the course of atrial fibrillation (Figure 12) as detailed below.
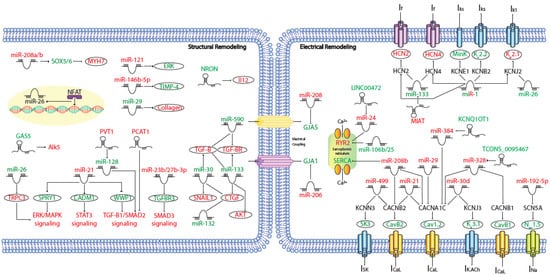
Figure 12. Schematic representation of the activated molecular program in atrial fibrillation. miRNAs and lncRNAs networks controlling structural and electrical remodelling in AF. (Red and green labels correspond with induced or repressed molecules in AF, respectively).
1.1. microRNAs and Electrical Disturbances in AF
IK1 disturbances are the most relevant ionic current changes underlying AF-induced electrical remodeling. As mentioned before, an increase in IK1, is a prominent feature of AF electrical remodeling and related to this process several miRNAs are involved. MiR-1 is a muscle-specific miRNA and the most abundantly expressed miRNA in both ventricles and atria [3][137]. It has been shown that down-regulation of miR-1 has proarrhythmic effect modulating IK1 through an up-regulation of potassium inwardly rectifying channel subfamily J member 2 (KCNJ2) expression [4][138]. Concomitantly, miR-26 is significantly reduced in AF patients compared to controls, leading to an IK1 increase, by direct targeting of KCNJ2. Nuclear factor of activated T cells (NFAT), a known actor in AF-associated remodeling, was found to negatively regulate miR-26 transcription [5][139]. Moreover, other groups have demonstrated that an up-regulation of miR-1 accelerates the shortening of the atrial effective refractory period (AERP) by targeting potassium voltage-gated channel subfamily E regulatory subunit 1 (KCNE1) and potassium voltage-gated channel subfamily B member 2 (KCNB2) [6][140]. Also, miR30d is significantly upregulated in cardiomyocytes from AF patients, whereas the mRNA and protein levels of KCNJ3/Kir3.1, postulated target of miR-30d, are markedly reduced, concomitant with a reduction of the acetylcholine-sensitive inward-rectifier K+ current (IK.ACh) [7][141]. Furthermore, miR-499 is up-regulated in permanent AF patients, this miRNA targets and down-regulates potassium calcium-activated channel subfamily N member 3 (KCNN3) and facilitates its recruitment into miRISCs, resulting in transcriptional repression of the small conductance calcium-activated potassium channel protein 3 (SK3) expression [8][142] (Table 13).
Table 13. AF associated microRNAs and their functional consequences in AF electrophysiology.
| Gene | Targets | Regulatory Role | AF Related Functional Consequences | Reference |
|---|---|---|---|---|
| miR-1 | KCNJ2 | increased IK1 current | increased membrane resting potential; increased AF vulnerability | [4][138] |
| KCNE1 | increased Iks current | decreased AERP; increased AF vulnerability | [6][140] | |
| KCNB2 | increased Iks current | decreased AERP; increased AF vulnerability | [6][140] | |
| HCN2 | increased expression | plausible increase in premature beats; increased AF vulnerability | [9][150] | |
| HCN4 | increased expression | plausible increase in premature beats; increased AF vulnerability | [9][150] | |
| miR-26 | KCNJ2 | increased IK1 current | increased membrane resting potential; increased AF vulnerability | [5][139] |
| miR-30d | KCNJ3 | reduced IK.Ach current | impaired calcium handling; increased AF vulnerability | [7][141] |
| miR-499 | KCNN3 | reduced SK3 expression | no direct evidences to AF pathophysiology | [8][142] |
| miR-192 | SCN5A | reduced Nav1.5 expression | no direct evidences to AF pathophysiology | [10][143] |
| miR-21 | CACNA1C | reduced Ica current | shortening APD; increased AF vulneratibility | [11][144] |
| CACNB2 | reduced Ica current | shortening APD; increased AF vulneratibility | [11][144] | |
| miR-29 | CACNA1C | reduced Ica current | no direct evidences to AF pathophysiology | [12][145] |
| miR-208ab | CACNA1C | reduced expression | potential impact in APD and thus on AF vulneratibility | [13][148] |
| CACNB2 | reduced expression | potential impact in APD and thus on AF vulneratibility | [13][148] | |
| GJA5 | indirect reduced expression | no direct evidences to AF pathophysiology | [14][152] | |
| ATP2A2 | reduced expression | no direct evidences to AF pathophysiology | [15][146] | |
| miR-328 | CACNA1C | reduced Ica current | shortening APD; increased AF vulneratibility | [16][147] |
| CACNB2 | reduced Ica current | shortening APD; increased AF vulneratibility | [16][147] | |
| miR-106b-25 | RYR2 | increased Ca++ release | increased pacing-induced AF vulnerability | [17][149] |
| miR-206 | GJA1 | reduced Cx43 expression | abnormal heart rate and PR interval; plausible link to AF | [18][151] |
Aside from IK1 remodeling, sodium channel (INa) density may be reduced in AF. In this context, it has been stablished that an upregulation of miR-192-5p in AF patients is corresponded to downregulation of SCN5A and Nav1.5 protein [10][143] (Table 13).
In addition, AF is characterized by a prominent downregulation of ICaL current and calcium handling remodeling. In this context, miR-21, whose expression levels are increased in myocytes isolated from chronic atrial fibrillation patients, decreases ICaL by downregulating calcium voltage-gated channel subunit alpha1 C (CACNA1C) and calcium voltage-gated channel auxiliary subunit beta 2 (CACNB2) [11][144]. Something similar happens with miR-29 and miR-30d, targeting CACNA1C [7][12][141,145]. Microarray screen in AF patients identified miR-208a and miR-208b, in particular, as the most significantly increased miRNAs in AF, miR-208b over-expression analysis showed that aberrant miR-208b levels reduce the expression and function of CACNA1C and CACNB2 [15][146]. Additionally, miR-328 has strong arrhythmogenic potential through a profound reduction of CACNA1C and CACNB1 and shortening of atrial action potential duration (APD) which augments the AF vulnerability [16][147]. As it has been previously mentioned, miR-499 is increased in AF patients and apart from regulating SK3 expression, it is also able to directly target CACNB2 [13][148] (Table 13). Meanwhile, miR-106b-25 cluster deficiency leads to atrial arrhythmogenesis via enhanced RyR2-mediated SR Ca2+-release [17][149] and miR-208b also reduces the expression and function of the sarcoplasmic reticulum-Ca2+ pump SERCA2 [15][146] (Table 13).
At the same time, it has been elucidated that the mRNA and protein expression levels of HCN2 and HCN4 channels increased with age, whereas miR-1 and miR-133 declined with age, implicating elevated HCN activity and reduced miR-1/133-mediated regulation of HCN expression in the pathogenesis of AF [9][150] (Table 13).
Besides ion channel function, to asses a proper electrical propagation between cardiomyocytes, it is necessary a correct regulation of connexin expression. In this context, miR-206 and miR-208 regulate gap junction protein alpha 1 (GJA1) and GJA5 respectively (Table 13). These miRNAs are increased in AF patients inducing cardiac arrhythmias [14][18][151,152], supporting the functional roles of these microRNAs in AF.
Several labs, including ours, demonstrated that Pitx2 deficiency disrupt microRNA expression that are linked to atrial arrhythmogenesis, a signaling path- way that also involves regulation of Wnt and Wnt- driven microRNAs expression, which is highly susceptible to alteration of cardiovascular risk factors such as hyperthyroidism, hypertension and redox homeostasis imbalance [19][20][21][22][130,153,154,155]. In sum, all these data support the notion that microRNAs play a fundamental role regulating key components that, if impaired, lead to AF electrical remodeling.
1.2. microRNAs and Structural Remodeling in AF
Structural remodeling is a long-lasting process that progressively affects myocytes and the myocardial interstitium, and takes place as early as the first days of atrial tachyarrhythmia onset [23][156]. miRNAs are involved in this process through gene regulation of proteins related to extracellular matrix deposition, apoptosis, and contractility.
Several miRNAs have been identified as potential participants in the regulation of the fibrotic remodeling occurring during AF. miR-21 represses sprouty RTK signaling antagonist 1 (SPRY1), a negative regulator of the extracellular signal-regulated kinase (ERK) pathway. In AF, ERK pathway is activated and promotes fibrosis indirectly through miR-21-induced SPRY1 downregulation [24][157]. Additionality, miR-21 also promotes cardiac fibrosis through the transcription factor signal transducer and activator of transcription 3 (STAT3) signaling pathway, by decreasing the expression of cell adhesion molecule 1 (CADM1) [25][158]. Finally, at the same time that miR-21 is up-regulated, WW domain containing E3 ubiquitin protein ligase 1 (WWP1) expression levels are down-regulated, promoting the activation of TGF-β1/Smad2 signaling pathway which endorses cardiac fibroblasts proliferation in AF patients [26][159]. By other hand, miR-23b and miR-27b overexpression enhance up-regulation of fibrosis-associated genes by targeting transforming growth factor beta receptor 3 (TGFBR3) [27][160] and posterior activation of SMAD3 signaling. Furthermore, miR-26 modulates Ca2+-permeable transient receptor potential canonical-3 (TRPC3) protein. miR-26 is down-regulated in AF, thus increasing TRPC3 expression which in turn stimulates fibroblast proliferation, differentiation, and activation [28][161]. miR-29 targets multiple extracellular matrix genes, including collagens, fibrillins and elastin, this miRNA is downregulated and its expression is inversely correlated with extracellular matrix protein levels and the development of AF [29][162]. In this context, miR-30a up-regulation reduces AF-induced myocardial fibrosis by targeting snail family transcriptional repressor 1 (SNAIL1) [30][163], whereas, miR-30c overexpression attenuates atrial fibrosis induced by TGF-β1, by targeting transforming growth factor beta receptor 2 (TGFβRII) [31][164], being both of them down-regulated in AF patients with an increase of fibrotic tissue. In addition, it has been demonstrated that miR-30, miR-133 and miR-132 regulate connective tissue growth factor (CTGF), which is a key molecule in the process of fibrosis, and collagen production, these miRNAs are down-regulated in AF patients promoting thus atrial fibrosis [32][33][165,166]. Also, it has been detected, that nicotine promotes AF by inducing atrial structural remodeling, through miR-133 and miR-590 downregulation and de-repression of TGF-β1 and TGFβRII [34][167]. Moreover, miR-146b-5p, matrix metallopeptidase 9 (MMP-9), involved in the degradation of extracellular matrix and formation of fibrosis, and collagen content were upregulated whereas tissue inhibitor of metalloproteinase 4 (TIMP-4) was downregulated in patients with AF [35][168]. Finally, AF patients showed a drastically increase of myosin heavy chain 7 (MYH7) protein levels, a hallmark of cardiac hypertrophy. It is suggested that the increased expression of miR-208a/b in AF contributes to high MYH7 protein levels via inhibiting the expression of SRY-box transcription factor 5 (SOX5) and SOX6, however the mechanistic implications of MYH7 in AF remain unclear [15][146].
Another layer of regulation of anatomical/structural components by miRNAs is apoptotic cell death, it has been demonstrated that miR-122 is up-regulated in AF patients, inhibiting ERK activation that leads apoptosis. In contrast, miR-133 has a cardioprotective role dependent on AKT serine/threonine kinase (AKT) signaling in control situation, inducing apoptosis in AF patients due to its down-regulation [36][37][169,170].
Apart from electrical and structural remodeling associated to AF, other miRNAs are involved in AF targeting related pathways, i.e., miR-21 modulates Phosphatase and Tensin Homolog (PTEN)/Phosphoinositide 3-kinase (PI3K) signaling pathway, signal transducer of transcription 3 (STAT3) and Smad7 promoting atrial fibrosis; miR-31 begets arrhythmia by depleting dystrophin and neuronal nitric oxide synthase (nNOS); miR-34a is upregulated in AF patients having an important role in the early electrophysiological changes and development of AF via regulation of the expression of Ankyrin-B (ANK 2); and finally, miR-199a down-regulation induces Sirtuin 1 (SIRT-1), a cardio-protective protein, as a compensatory mechanism to inhibit the process of oxidative stress which contributes to the pathogenesis of AF [38][39][40][41][42][43][171,172,173,174,175,176].
All these data support the role of miRNAs in AF pathophysiology. Functional studies targeting miRNAs are necessary to study the therapeutic potential of these molecules in treating cardiovascular disease, although there are multiple concerns as to the safety of miRNA therapeutics, as miRNAs’ ability to target multiple pathways within the target tissue or in different organs, with further research being needed to confirm the safety of miRNAs.
2. lncRNAs and Atrial Fibrillation
Long non-coding RNAs (lncRNAs) are currently defined as noncoding RNAs large that 200 nucleotides. LncRNAs constitute a widely diverse group of non-coding RNAs with structural similarities to protein-coding RNAs but with no or limited capacity to code for proteins. LncRNAs display a variety of transcriptional and post-transcriptional functions, such as scaffold platform, modulation of epigenetic factors and protein translation among others. Some of these studies were performed in lone AF patients [44][45][46][179,180,181], whereas in others valvular heart disease [47][177] or rheumatic valve disease [45][48][49][178,180,182] was also concurring. In most cases, either right or left atrial appendages were analysed [42][43][44][45][47][48][175,176,177,178,179,180], but in some cases right and left atrial samples were pooled together [44][45][46][179,180,181], while in other blood samples [45][50][180,183] or epicardial adipose tissue [51][52][184,185] were analysed. Given the wide variability of biological conditions studied, it is then not surprising that comparative analyses of these lncRNA transcriptomic analyses revealed no common AF signature [53][186]. Importantly, concurrent analyses of differentially expressed mRNAs and/or microRNAs and more recently circular RNAs (circRNAs) have provided additional insights into the plausible gene regulatory networks involved in AF [47][48][51][54][177,178,184,187]. Unfortunately, to date, only the lncRNA fingerprints have been provided and functional assays are scarcely reported.
At present, assays of the functional role of lncRNAs have only been reported in experimental animal models or in in vitro assays. Several of these studied reported a functional role of lncRNAs modulating fibrosis [55][56][57][58][188,189,190,191], ion channel function [59][60][61][62][63][64][192,193,194,195,196,197], and energy metabolism [65][198] as detailed below.
2.1. lcnRNAs in AF Structural Remodelling
Four distinct studies have reported the functional role of lncRNA in AF fibrosis. Cao et al. [56][189] reported increased PVT1 lncRNA expression in human AF atrial biopsies and furthermore they demonstrate a role for PVT1 enhancing atrial fibroblast proliferation and collagen deposition by sponging miR-128-3p that in turn promoted Tgfb/Smads signaling. Additionally, indirect reports of the functional role of lncRNA in atrial fibrosis have been reported by Lu et al. [57][190] and Chen et al. [58][191]. Lu et al. [57][190] reported significantly reduced GAS5 expression in the right atrial appendage (RAA) of AF patients while GAS5 manipulation in AC16 cells, controlled cell growth by modulating ALK5 expression. Chen et al. [58][191] demonstrated increased PCAT-1 expression in right atrial appendage of AF patients. Knockdown of PCAT-1 inhibited proliferation in AC16 cell by modulating transforming growth factor-β1 (TGF-β1). Finally, Sun et al. [60][193] demonstrated that NRON overexpression suppressed, while silencing facilitated, angiotensin II (Ang II)-induced inflammatory response in primary cultured atrial myocytes. Chromatin immunoprecipitation (ChIP) assays showed that nuclear factor of activated T cell 3 (NFATc3) was recruited to the promoter region of interleukin 12 activating its expression in atrial myocytes. Collectively, the authors demonstrated that lncRNA NRON alleviates atrial fibrosis through suppression of M1 macrophages activated by atrial myocytes.
2.2. lcnRNAs in AF Electrical Remodelling
Several reports provided evidence on the functional role of lncRNAs modulating the cardiac electrophysiological properties in AF, particularly those related to calcium regulation and handling. Shen et al. [61][194] reported that KCNQ1OT1 is up-regulated in AngII-treated mice as well as in an experimental AF mouse model. The authors demonstrate that KCNQ1OT1 regulates CACNA1C by sponging miR-384. KCNQ1OT1 manipulation modulate distinct electrophysiological parameters such as the effective refractory period and the interatrial conduction and KCNQ1OT1 silencing diminishes the incidence of AF and AF episodes in AngII-treated mice (Table 24).
Table 24.
AF associated lncRNAs and their functional consequences in AF electrophysiology.
| Gene | Targets | Regulatory Role | AF Related Functional Consequences | Reference |
|---|---|---|---|---|
| KCNQ1OT1 | CACNA1C | miR-384 sponge | impaired AERP and the interatrial conduction; increased AF vunerability | [61][194] |
| TCONS_00075467 | CACNA1C | miR-328 sponge | reduced ICa and shortened APD and AERP; increased AF vunerability | [59][192] |
| LINC00472 | unknown | miR24/JP2/RyR2 | no direct evidences to AF pathophysiology | [62][195] |
| TCONS_00202959 | unknown | unknown | shortened AERP and increased AF vunerability | [63][196] |
| TCONS_00032546 | unknown | unknown | shortened AERP and increased AF vunerability | [64][197] |
| TCONS_00026102 | unknown | unknown | increased AERP and prevented AF inducibility | [64][197] |
| MIAT | unknown | miR-133 sponge | increased AERP and prevented AF inducibility | [65][198] |
Li et al. [59][192] reported the lncRNA expression profiles of right atria in AF and non-AF rabbit models and identified 1220 differentially expressed transcripts. TCONS_00075467 was selected for further exploration. In vivo silencing of TCONS_00075467 leading to shortening of the atrial effective refractory period and the L-type calcium current and action potential duration were decreased in vitro. Additionally, the authors demonstrated that TCONS_00075467 sponge miRNA-328 both in vitro and in vivo thus regulating CACNA1C. (Table 24).
More recently, it has been reported that AF patients displayed increased miR-24 and reduced LINC00472 expression, while LINC00472 DNA promoter methylation was also increased. Functional evidence demonstrated that miR-24 can negatively regulate LINC00472 and JP2 expression, and thus LINC00472 could regulate the progression of AF via modulating the LINC00472/miR-24/JP2/RyR2 signaling pathway [62][195] (Table 24).
The modulative effects of lncRNAs on autonomic neural function and myocardial functions in atrial fibrillation rat model have been also recently investigated [63][196]. These authors show that over-expression of TCONS_00202959 in an experimental rat AF model enhances the atrial effective refractory period and diminishes the AF induction rate. However, the precise molecular mechanisms by which AERP is decreased remains unclear (Table 24). Similarly, Wang et al. [64][197] reported the fat pads lncRNA profile in an AF experimental canine model. These authors reported 166 down-regulated and 410 up-regulated (576 differentially expressed lncRNA) lncRNAs and they further underwent to dissect the functional role of two of these differentially expressed lncRNAs, TCONS_00032546 and TCONS_00026102, by in vivo silencing, leading to a significant shortening or prolongation the atrial effective refractory period, and thereby these lncRNAs increased or prevented AF inducibility, respectively (Table 24).
More recently it has been reported complementary expression patterns for MIAT and miR-133a-3p in an experimental AF rat model as well as in peripheral blood leukocyte samples of AF patients [65][198]. These authors further demonstrated that miR-133a-3p directly regulates by MIAT. MIAT knockdown significantly reverted AF, increasing atrial effective refractory period and thus reducing the duration of AF. Importantly, cardiomyocyte apoptosis and atrial fibrosis were also reduced (Table 24).
A functional role for lncRNAs in cardiomyocyte metabolism was reported by Chen et al. [66][199]. These authors performed a microarray analysis using pulmonary vein myocardium and the surrounding myocardium and compared to LA appendage leading to the identification of 94 differentially expressed lncRNAs, among which AK055347 was one of lncRNAs most significantly altered. Experimental manipulation of this lncRNA further demonstrate a role in mitochondrial energy production. In sum, these data support an emerging functional role for lncRNAs in AF.
3. DNA Methylation and Atrial Fibrillation
DNA methylation is a pre-transcriptional modification, which is able to change transcriptional process, by the addition of methyl groups to specific nucleotides of the DNA. This procedure causes inactive gene expression due to the fact that the methyl binding protein prevents the transcriptional factor from binding to DNA and thus it proceed to the next step [67][68][69][200,201,202]. It is commonly believed that hypomethylation in diseases is more frequent than hypermethylation [70][203].
However, in AF context, global DNA methylation levels are significantly increased in AF patients, having a positive correlation with age [69][202]. Furthermore, it has been demonstrated that DNA methylation plays an important role in the maintenance of cardiac fibrosis, where DNA methyltransferases 3A (DNMT3A) likely plays an essential role in Ras association domain family member 1A (RASSF1A) mediated up-regulation of ERK1/2 [71][72][204,205]. Moreover, heart failure induces Pitx2c promoter hypermethylation and Angiotensin II may contribute to the hypermethylation in heart failure [73][206]. In addition, tumor necrosis factor- α (TNF-α) decreases SERCA2 expression via DNMT1 which induces promoter methylation in cardiomyocytes [74][207]. Emelia’s lab has identified two CpG sites significantly associated with prevalent AF, and five CpGs associated with incident AF, and fourteen previously reported genome-wide significant AF-related SNP were each associated with at least one CpG site; being the most significant association rs6490029 at the CUX2 locus and cg10833066 [75][208]. Recently studies have shown that KIF15 methylation may play important role in the pathogenesis of AF through the regulation of the expression of proteasome 26S subunit ATPase 3 (PSMC3), tubulointerstitial nephritis antigen (TINAG), and nudix hydrolase 6 (NUDT6) [76][209].
These results suggest that DNA methylation might represent an important molecular process that is able to link genetic variations with AF susceptibility. To date, only a few studies have investigated differential DNA methylation as a predictor biomarker at specific candidate loci that were previously associated with AF.
4. Histone Modifications and HDACs in AF
Histone modification represent an important mechanism of epigenetic regulation. The N-terminal of histones can undergo distinct post-transcriptional modifications and the most common modifications include phosphorylation, acetylation, methylation, and ubiquitination, but others occur as well [77][210]. Such post-transcriptional modifications thus play important biological roles in a wide array of cellular processes including cell cycle and metabolism control, DNA repair and particularly important on gene transcription. To date, only post-transcriptional modification by acetylation has been reported in association to AF.
Histone acetylation, modulated by histone acetyltransferases (HATs) is normally associated to open chromatin configurations and thus to active gene transcription while histone deacetylation, catalysed by distinct classes of histone deacetylases (HDACs) is linked to gene silencing [68][201]. Currently, the ouresearchers' understanding of the functional impact of HDAC in histone modification in the setting of AF remains largely unexplored. However, HDAC, besides post-transcriptionally regulating histones and the nuclear chromatin, can also be translocated into the cytoplasm modulating acetylation and deacetylation of other proteins [78][79][80][211,212,213]. In this context, emerging evidence is demonstrating a pivotal role of HDACs influencing post-transcriptional regulation of distinct proteins in cardiomyocytes in the context of AF [81][82][214,215], particularly on cytoskeletal [80][213] and conductive proteins [83][216], while their role in contractile and ion channels remains unclear [84][217]. Additionally, HDAC inhibition can significantly block or halt AF progression [83][85][86][87][88][216,218,219,220,221], further supporting the important role of HDAC in AF, yet the molecular mechanisms remain to be further explored.