Metabolic activities of cheese microbiota play a crucial role in the development of cheese typicity. For geographical origin authentication, microbiota fingerprinting is therefore of high interest as traditional and artisanal cheeses are produced with a more diversified microbiota associated with the cheese-making process (e.g., use of raw milk, starter, brine, equipment and materials, and ripening rooms). While cheese microbial diversity was traditionally investigated using culture-dependent methods, hence overlooking unculturable or subdominant species, nowadays, culture-independent methods (HTS) have unravelled this diversity and provided further means to connect microbiota composition to cheese quality and typicity, but also origin. This success is due to the availability of new sequencing platforms, bioinformatic pipelines, and a continuous decrease in cost. While complete microbial genome sequencing (i.e. metagenomics) can be used, metabarcoding (also known as metagenetics and corresponding to the selective amplification of taxonomically relevant followed by NGS sequencing) is the most widely reported in the scientific literature. Herein we report recent scientific literature about metabarcoding analysis of cheese microbiota, highlighting what factors contribute to its formation and how it could be used to authenticate cheese origin.
- cheese
- geographical origin
- authentication
- next-generation sequencing
1. Introduction
To perform DNA metabarcoding, cheese samples are first homogenized, then total DNA is most frequently extracted using commercial kits, ad hoc protocols or a combination thereof [1][2]. Hypervariable regions of taxonomically relevant genes (e.g., 16S rDNA for bacteria and archaea, ITS, 18S rDNA, 26S rDNA for fungi) are amplified by PCR reactions, while a second amplification step tags amplicons with specific DNA fragments –barcodes- and dedicated adapters for the final sequencing step using next-generation technologies (e.g., Illumina, Pacbio, Iontorrent, or Nanopore). Typically, 16S rDNA and ITS (internal transcribed spacer) markers, targeting bacteria and fungi, respectively, are employed to generate compositional data describing microbial taxa and their relative abundance in cheese microbial communities. After sequencing, two complementary but different ways can be used for amplicon clustering from quality-checked data, namely, operational taxonomic units (OTUs) and amplicon sequencing variants (ASVs) [3]. Subsequently, taxonomic assignment is performed using a specific classifier tool (BLAST, RDP, UCLUST, SortMeRNA) against various reference databases, such as Greengenes, SILVA, and UNITE [4][5]. Generally, clustered OTUs/ASVs are analyzed from the phylum to the genus level since they can be less precise at the species level [6].
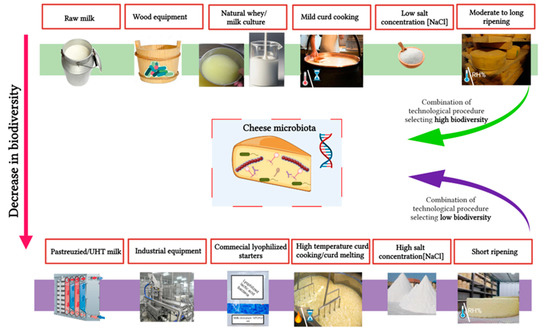
12. Main Factors Affecting Cheese Microbial Diversity
23. Climatic and Environmental Condition
34. Cheese Ripening
| Advantages | Reference | Limitations | Reference |
|---|---|---|---|
| Time- and cost-effective processing of large sample numbers | [30] | Analyses could be biased by sample processing, DNA extraction methods, and equimolar library preparation | [31] |
| Consolidated pipeline for data analysis | [32] | PCR amplification steps include errors, e.g., PCR specificity and variation of 16S rRNA copy number per genome | [33] |
| Identification of taxonomic groups associated with typical flavor and cheese-making technology | [34] | Under- or over-estimation of microbial community diversity | [32][35] |
| Allows improvement of cheese-making to ensure safety while preserving typicity | [7] | Lack of absolute abundance | [36][37] |
| Evaluation of core microbiome describing facility-associated microbial groups | [38][39] | Limited and uneven taxonomic resolutions | [40][41] |
| May pinpoint new biotypes | [17] | DNA amplicon sequencing typically does not discriminate between live and dead microorganisms (except if DNA stains such as propidium monoazide are used) | [42] |
References
- Benjamin E. Wolfe; Julie E. Button; Marcela Santarelli; Rachel J. Dutton; Cheese Rind Communities Provide Tractable Systems for In Situ and In Vitro Studies of Microbial Diversity. Cell 2014, 158, 422-433, 10.1016/j.cell.2014.05.041.
- Ilhan Cem Duru; Pia Laine; Margarita Andreevskaya; Lars Paulin; Soila Kananen; Soile Tynkkynen; Petri Auvinen; Olli-Pekka Smolander; Metagenomic and metatranscriptomic analysis of the microbial community in Swiss-type Maasdam cheese during ripening. International journal of food microbiology 2018, 281, 10-22, 10.1016/j.ijfoodmicro.2018.05.017.
- Sandra Reitmeier; Thomas C. A. Hitch; Nicole Treichel; Nikolaos Fikas; Bela Hausmann; Amanda E. Ramer-Tait; Klaus Neuhaus; David Berry; Dirk Haller; Ilias Lagkouvardos; et al.Thomas Clavel Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Communications 2021, 1, 1-12, 10.1038/s43705-021-00033-z.
- Christian Quast; Elmar Pruesse; Pelin Yilmaz; Jan Gerken; Timmy Schweer; Pablo Yarza; Jörg Peplies; Frank Oliver Glöckner; The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 2012, 41, D590-D596, 10.1093/nar/gks1219.
- Henrik Nilsson; Karl-Henrik Larsson; Andy F S Taylor; Johan Bengtsson-Palme; Thomas Stjernegaard Jeppesen; Dmitry Schigel; Peter Kennedy; Kathryn Picard; Frank Oliver Glöckner; Leho Tedersoo; et al.Irja SaarUrmas KõljalgKessy Abarenkov The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Research 2018, 47, D259-D264, 10.1093/nar/gky1022.
- Orna Mizrahi-Man; Emily Davenport; Yoav Gilad; Taxonomic Classification of Bacterial 16S rRNA Genes Using Short Sequencing Reads: Evaluation of Effective Study Designs. Plos One 2013, 8, e53608, 10.1371/journal.pone.0053608.
- Kamimura, B.A.; De Filippis, F.; Sant’Ana, A.S.; Ercolini, D. Large-Scale Mapping of Microbial Diversity in Artisanal Brazilian Cheeses. Food Microbiol. 2019, 80, 40–49.
- Sant’Anna, F.M.; Wetzels, S.U.; Cicco, S.H.S.; Figueiredo, R.C.; Sales, G.A.; Figueiredo, N.C.; Nunes, C.A.; Schmitz-Esser, S.; Mann, E.; Wagner, M.; et al. Microbial Shifts in Minas Artisanal Cheeses from the Serra Do Salitre Region of Minas Gerais, Brazil throughout Ripening Time. Food Microbiol. 2019, 82, 349–362.
- Li, J.; Zheng, Y.; Xu, H.; Xi, X.; Hou, Q.; Feng, S.; Wuri, L.; Bian, Y.; Yu, Z.; Kwok, L.-Y.; et al. Bacterial Microbiota of Kazakhstan Cheese Revealed by Single Molecule Real Time (SMRT) Sequencing and Its Comparison with Belgian, Kalmykian and Italian Artisanal Cheeses. BMC Microbiol. 2017, 17, 13.
- Lucchini, R.; Cardazzo, B.; Carraro, L.; Negrinotti, M.; Balzan, S.; Novelli, E.; Fasolato, L.; Fasoli, F.; Farina, G. Contribution of Natural Milk Culture to Microbiota, Safety and Hygiene of Raw Milk Cheese Produced in Alpine Malga. Ital. J. Food Saf. 2018, 7, 6967.
- Zago, M.; Bardelli, T.; Rossetti, L.; Nazzicari, N.; Carminati, D.; Galli, A.; Giraffa, G. Evaluation of Bacterial Communities of Grana Padano Cheese by DNA Metabarcoding and DNA Fingerprinting Analysis. Food Microbiol. 2021, 93, 103613.
- Turri, F.; Cremonesi, P.; Battelli, G.; Severgnini, M.; Brasca, M.; Gandini, G.; Pizzi, F. High Biodiversity in a Limited Mountain Area Revealed in the Traditional Production of Historic Rebel Cheese by an Integrated Microbiota–Lipidomic Approach. Sci. Rep. 2021, 11, 10374.
- Dalmasso, A.; Soto del Rio, M.d.l.D.; Civera, T.; Pattono, D.; Cardazzo, B.; Bottero, M.T. Characterization of Microbiota in Plaisentif Cheese by High-Throughput Sequencing. LWT-Food Sci. Technol. 2016, 69, 490–496.
- Xue, J.; Yang, Y.; Wang, Z.; Guo, Y.; Shao, Y. Bacterial Diversity in Chinese Rushan Cheese From Different Geographical Origins. Front. Microbiol. 2018, 9, 1920.
- Piqueras, J.; Chassard, C.; Callon, C.; Rifa, E.; Theil, S.; Lebecque, A.; Delbès, C. Lactic Starter Dose Shapes S. Aureus and STEC O26:H11 Growth, and Bacterial Community Patterns in Raw Milk Uncooked Pressed Cheeses. Microorganisms 2021, 9, 1081.
- Johnson, J.; Curtin, C.; Waite-Cusic, J. The Cheese Production Facility Microbiome Exhibits Temporal and Spatial Variability. Front. Microbiol. 2021, 12, 644828.
- Kamilari, E.; Anagnostopoulos, D.A.; Papademas, P.; Kamilaris, A.; Tsaltas, D. Characterizing Halloumi Cheese’s Bacterial Communities through Metagenomic Analysis. LWT 2020, 126, 109298.
- Aldrete-Tapia, A.; Escobar-Ramírez, M.C.; Tamplin, M.L.; Hernández-Iturriaga, M. High-Throughput Sequencing of Microbial Communities in Poro Cheese, an Artisanal Mexican Cheese. Food Microbiol. 2014, 44, 136–141.
- Gobbetti, M.; Di Cagno, R.; Calasso, M.; Neviani, E.; Fox, P.F.; De Angelis, M. Drivers That Establish and Assembly the Lactic Acid Bacteria Biota in Cheeses. Trends Food Sci. Technol. 2018, 78, 244–254.
- Porcellato, D.; Skeie, S.B. Bacterial Dynamics and Functional Analysis of Microbial Metagenomes during Ripening of Dutch-Type Cheese. Int. Dairy J. 2016, 61, 182–188.
- Cardinali, F.; Ferrocino, I.; Milanović, V.; Belleggia, L.; Corvaglia, M.R.; Garofalo, C.; Foligni, R.; Mannozzi, C.; Mozzon, M.; Cocolin, L.; et al. Microbial Communities and Volatile Profile of Queijo de Azeitão PDO Cheese, a Traditional Mediterranean Thistle-Curdled Cheese from Portugal. Food Res. Int. 2021, 147, 110537.
- Ceugniez, A.; Taminiau, B.; Coucheney, F.; Jacques, P.; Delcenserie, V.; Daube, G.; Drider, D. Fungal Diversity of “Tomme d’Orchies” Cheese during the Ripening Process as Revealed by a Metagenomic Study. Int. J. Food Microbiol. 2017, 258, 89–93.
- Biolcati, F.; Bottero, M.T.; Dalmasso, A. Microbiological Analysis of the Robiola Di Roccaverano Cheese by Means of Traditional Culturing Methods. Ital. J. Food Saf. 2019, 8.
- Montel, M.-C.; Buchin, S.; Mallet, A.; Delbes-Paus, C.; Vuitton, D.A.; Desmasures, N.; Berthier, F. Traditional Cheeses: Rich and Diverse Microbiota with Associated Benefits. Int. J. Food Microbiol. 2014, 177, 136–154.
- Martín, J.F.; Coton, M. Blue cheese: Microbiota and fungal metabolites. In Fermented Foods in Health and Disease Prevention; Academic Press: San Diego, CA, USA, 2017; pp. 275–303.
- Coton, E.; Coton, M.; Hymery, N.; Mounier, J.; Jany, J.-L. Penicillium Roqueforti: An Overview of Its Genetics, Physiology, Metabolism and Biotechnological Applications. Fungal Biol. Rev. 2020, 34, 59–73.
- Gillot, G.; Jany, J.-L.; Poirier, E.; Maillard, M.-B.; Debaets, S.; Thierry, A.; Coton, E.; Coton, M. Functional Diversity within the Penicillium Roqueforti Species. Int. J. Food Microbiol. 2017, 241, 141–150.
- Garnier, L.; Valence, F.; Mounier, J. Diversity and Control of Spoilage Fungi in Dairy Products: An Update. Microorganisms 2017, 5, 42.
- Mounier, J.; Coton, M. Kluyveromyces spp. In Encyclopedia of Dairy Sciences; McSweeney, P.L.H., McNamara, J.P., Eds.; Elsevier: San Diego, CA, USA, 2022; Volume 4, pp. 569–574.
- Lundin, D.; Severin, I.; Logue, J.B.; Östman, Ö.; Andersson, A.F.; Lindström, E.S. Which Sequencing Depth Is Sufficient to Describe Patterns in Bacterial α- and β-Diversity? Sequencing Depth in Diversity Research. Environ. Microbiol. Rep. 2012, 4, 367–372.
- Rubin, B.E.R.; Sanders, J.G.; Hampton-Marcell, J.; Owens, S.M.; Gilbert, J.A.; Moreau, C.S. DNA Extraction Protocols Cause Differences in 16S RRNA Amplicon Sequencing Efficiency but Not in Community Profile Composition or Structure. MicrobiologyOpen 2014, 3, 910–921.
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An Independent Evaluation of Microbiome Sequence Error-Correction Approaches. PeerJ 2018, 6, e5364.
- Kleerebezem, R.; Stouten, G.; Koehorst, J.; Langenhoff, A.; Schaap, P.; Smidt, H. Experimental Infrastructure Requirements for Quantitative Research on Microbial Communities. Curr. Opin. Biotechnol. 2021, 67, 158–165.
- Unno, R.; Suzuki, T.; Matsutani, M.; Ishikawa, M. Evaluation of the Relationships Between Microbiota and Metabolites in Soft-Type Ripened Cheese Using an Integrated Omics Approach. Front. Microbiol. 2021, 12, 681185.
- Hugerth, L.W.; Andersson, A.F. Analysing Microbial Community Composition through Amplicon Sequencing: From Sampling to Hypothesis Testing. Front. Microbiol. 2017, 8, 1561.
- Piwosz, K.; Shabarova, T.; Pernthaler, J.; Posch, T.; Šimek, K.; Porcal, P.; Salcher, M.M. Bacterial and Eukaryotic Small-Subunit Amplicon Data Do Not Provide a Quantitative Picture of Microbial Communities, but They Are Reliable in the Context of Ecological Interpretations. mSphere 2020, 5, e00052-20.
- Yeh, Y.-C.; Needham, D.M.; Sieradzki, E.T.; Fuhrman, J.A. Taxon Disappearance from Microbiome Analysis Reinforces the Value of Mock Communities as a Standard in Every Sequencing Run. mSystems 2018, 3, e00023-18.
- Bokulich, N.A.; Mills, D.A. Facility-Specific “House” Microbiome Drives Microbial Landscapes of Artisan Cheesemaking Plants. Appl. Environ. Microbiol. 2013, 79, 5214–5223.
- Calasso, M.; Ercolini, D.; Mancini, L.; Stellato, G.; Minervini, F.; Di Cagno, R.; De Angelis, M.; Gobbetti, M. Relationships among House, Rind and Core Microbiotas during Manufacture of Traditional Italian Cheeses at the Same Dairy Plant. Food Microbiol. 2016, 54, 115–126.
- Brumfield, K.D.; Huq, A.; Colwell, R.R.; Olds, J.L.; Leddy, M.B. Microbial Resolution of Whole Genome Shotgun and 16S Amplicon Metagenomic Sequencing Using Publicly Available NEON Data. PLoS ONE 2020, 15, e0228899.
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S RRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029.
- Afshari, R.; Pillidge, C.J.; Dias, D.A.; Osborn, A.M.; Gill, H. Cheesomics: The Future Pathway to Understanding Cheese Flavour and Quality. Crit. Rev. Food Sci. Nutr. 2020, 60, 33–47.