Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Julio Camarero and Version 2 by Dean Liu.
The selective disruption of protein–protein interactions remains challenging, as the interacting surfaces are relatively large and flat. However, highly constrained polypeptide-based molecular frameworks with cell-permeability properties, such as the cyclotide scaffold, have shown great promise for targeting those biomolecular interactions.
- cyclotides
- drug design
- backbone cyclized polypeptides
- protein–protein interactions
1. Introduction
Disruption of pharmacologically relevant protein–protein interactions (PPIs) remains a challenging task [1][2][3][1,2,3]. This is primarily due to the large and relatively flat binding surfaces involved in most PPIs. The most challenging molecular targets are those involving intracellular PPIs, which also require the therapeutic agent to cross the cell membrane efficiently [4][5][4,5].
Generally, rwesearchers can consider two major structural types of therapeutic agents, small molecules and protein-based compounds, known as biologicals. Small molecules are small in molecular size (≤100 atoms) and usually show good pharmacological properties, such as cell permeability and stability. However, small molecules only provide a modest overall surface area available for interacting with the protein target. This has made quite challenging the identification of small molecules able to efficiently disrupt PPIs [6][7][6,7].
The use of polypeptide-based molecules, on the other hand, has provided efficient therapeutic tools to modulate PPIs with high specificity and selectivity [8]. For example, therapeutic monoclonal antibodies can target extracellular protein domains in a remarkably efficient fashion [9][10][9,10]. Antibodies, however, are expensive to produce, show low tissue penetration, are unable to reach intracellular targets, and cannot be delivered orally. These limitations have led to exploring alternative polypeptide-based scaffolds as a potential source of protein-based therapeutic leads [11][12][13][14][15][16][17][18][11,12,13,14,15,16,17,18].
The use of highly constrained polypeptides and able to cross membranes has recently received special attention for developing de novo stable polypeptide-based therapeutics [11][12][19][11,12,19]. Among the different highly-constrained peptide-based scaffolds, the cyclotide family has emerged as a fascinating family of medium-sized and Cys-rich plant-derived backbone-cyclized polypeptides (≈30–40 amino acids long). Cyclotides possess a stabilizing core formed by three disulfide bonds forming a Cys-knotted arrangement (Figure 1) [19]. This Cys-knotted backbone-cyclized (or circular) topology confers cyclotides with unusual characteristics such as remarkable stability to thermal/chemical denaturation and proteolytic degradation [20]. These unusual features have made cyclotides ideal tools for developing novel peptide-based therapeutic leads (see some recent reviews on the topic [11][21][22][23][24][25][26][11,21,22,23,24,25,26]. Due to their relatively small sizes, cyclotides can be chemically produced by standard solid-phase peptide synthesis (SPPS) methods and can also be produced by heterologous expression in different types of cells using standard expression vectors (see a recent review on the biological and chemical production of cyclotides [27]). Another intriguing property of cyclotides is that some of them can cross the cellular membranes of mammalian cells through endocytic mechanisms [28][29][28,29] and be able to modulate PPIs in vitro and in vivo [5]. Some naturally occurring cyclotides have also shown biological activity when given orally hence showing some oral bioavailability [21][30][31][21,30,31]. The first cyclotide discovered in plants, kalata B1 (Figure 1), was employed in traditional medicine as an effective uterotonic agent when given orally [20]. Other kalata B1-derived cyclotides have also been shown to possess biological activity when dosed orally [30][31][30,31].
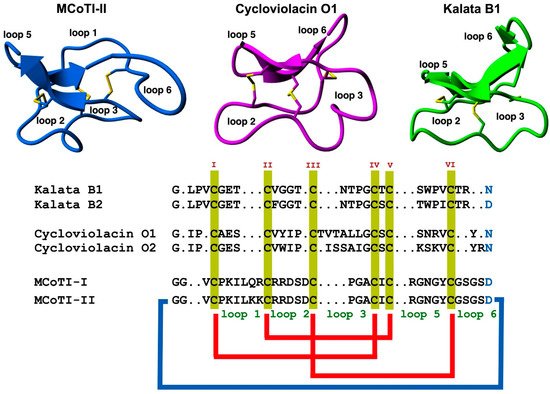
Figure 1. Structure and sequence alignment of cyclotides from the trypsin inhibitor (MCoTI-II, pdb: 1IB9) [32], bracelet (cycloviolacin O1, pdb: 1NBJ) [33], and Möbius (kalata B1, pdb: 1NB1) [33], subfamilies. Loops connecting the different Cys residues are designated with Arabic numerals, and the six Cys residues involved in the Cys-knot are labeled with roman numerals. Conserved Asp/Asn (required for backbone cyclization in nature) and Cys residues are marked in blue and yellow, respectively. Molecular graphics were created using Yasara (www.yasara.org) (accessed on 16 August 2022). Figure adapted from references [11][23][11,23].
2. Structure
Cyclotides consist of a backbone-cyclized polypeptide with six Cys residues that form a Cys-knotted structure. Naturally-occurring cyclotides are medium-sized polypeptides containing from 27 to 37 amino acids (Figure 1), although larger engineered cyclotides have also been reported [5][33][34][5,33,34]. Loop 6 seems to be the most tolerant for the insertion of large sequences using molecular grafting techniques, tolerating the insertion of sequences from 14 to up to 25 residues [5][34][5,34]. This loop also seems to tolerate well the introduction of isopeptide bonds without affecting the folding and stability of the resulting engineered cyclotide [35].
As mentioned earlier, the six Cys residues on the cyclotide scaffold form three interlocked disulfides on a well-defined Cys-knot arrangement, with disulfide CysIII-CysVI running through the ladder arrangement formed by disulfides CysI-CysIV and CysII-CysV (Figure 1 and Figure 2A). This highly constrained topology, known as the cyclic cystine knot (CCK) motif, makes the cyclotide backbone extremely rigid and compact [36]. This explains their unusual stability to thermal/chemical denaturation as well as proteolytical degradation that is characteristic of naturally occurring as well as engineered cyclotides [37][38][37,38].
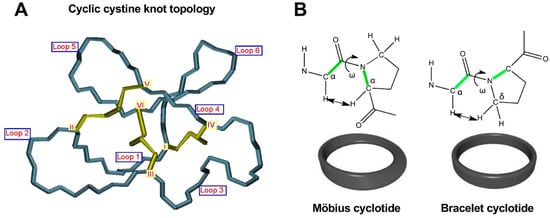
Figure 2. Detailed structural features of the cyclic cystine knot (CCK) topology observed in all naturally-occurring cyclotides. (A) Three-dimensional structure of the CCK architecture topology and the connecting loops found in cyclotides. Cys residues are labeled with roman numerals and loops connecting the different Cys residues are identified with Arabic numerals. (B) Cyclotides from the Möbius subfamily have a cis-Pro residue located in loop 5 that is responsible for inducing a local 180° backbone twist. This feature is absent in cyclotides from the other two subfamilies. Figure adapted from reference [26].
These properties were initially showcased on the first naturally occurring cyclotide, kalata B1, which was isolated and identified in the late 1960s by Gran while studying the traditional remedy employed by the indigenous people in central Africa use to accelerate childbirth [39]. This traditional medicine was prepared by boiling parts of the plant Oldelandia affinis (Rubiaceae family) to prepare the tea extract used as a remedy [40]. These early findings highlight the remarkable stability of the cyclotide scaffold that was biologically active even after being extracted by boiling water and providing uterotonic activity when dosed orally.
Since the discovery of the first cyclotide, more cyclotides have been isolated from other plant families [41]. Naturally occurring cyclotides have been mainly classified into three subfamilies, the Möbius, bracelet, and trypsin inhibitor cyclotide subfamilies [42]. Although all the cyclotides from the different subfamilies share the same CCK topology, they show differences in the size and sequence of the different loops (Figure 1). An additional structural difference between the cyclotides from the Mobius and bracelet subfamilies is that Möbius cyclotides have a cis-Pro bond at loop 5 while bracelet cyclotides do not have it (Figure 2B) [33].
Even though bracelet cyclotides are by far the most abundant in nature, where they make up around 70% of all the know cyclotides thus far, they are very difficult to fold in vitro [43]. A recent report, however, has shown that introducing a single point mutation in loop 2, replacing Ile11 with either a Gly or Leu residue (Figure 1), can substantially increase the folding yield in bracelet cyclotides [44]. This was demonstrated in several bracelet cyclotides. This approach was successfully used to synthesize mirror image enantiomers and used quasi-racemic crystallography, allowing to elucidate of the first crystal structures of bracelet cyclotides containing an Ile residue in loop 2 [44]. This study should offer an alternative and efficient approach to obtaining bracelet cyclotides, facilitating easy access to their three-dimensional structures and providing a basis for further study of cyclotide structure and function and their future use as drug design scaffolds [44].
The trypsin inhibitor subfamily contains a relatively smaller number of cyclotides isolated from the seeds of several Momordica spp. plants (Cucurbitaceae family) [45][46][47][45,46,47]. These cyclotides do not share significant sequence homology with members from the Möbius and bracelet subfamilies beyond the CCK topology. As its name indicates, cyclotides from this subfamily are extremely potent trypsin inhibitors (Ki ≈ 20 pM) [48]. Cyclotides of this family show high sequence homology with squash trypsin inhibitors, which also contain a Cys-knotted structure, although they are not backbone cyclized, and sometimes are referred to as cyclic knottins [49].
New naturally occurring cyclotides with sequences rich in positively charged Lys residues have also been recently isolated from two plants from the Violaceae family in Australia (49). Unfortunately, so far, there is no information available on their chemical synthesis, making it difficult to evaluate their real potential as molecular frameworks in the design of novel peptide-based therapeutics.
3. Biosynthesis
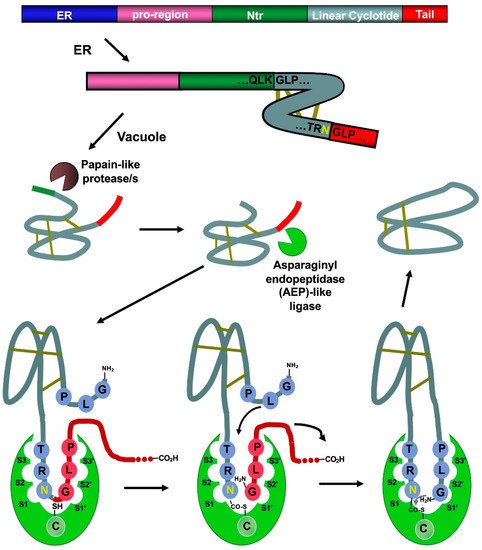
Naturally occurring cyclotides are produced by enzymatic processing from ribosome-produced precursor proteins (Figure 3). Many of these precursors are encoded in genes containing multiple copies of the same or different cyclotide sequences [50]. For example, the first dedicated genes to produce cyclotides were isolated from the cyclotide-producing plant Oldelandia affinis (Rubiaceae family), which is the natural source for cyclotide kalata B1 [51][52][51,52]. The genome analysis in other cyclotide-producing plants from different families has also allowed the identification of similar genes involved in the bioproduction of cyclotides [19][46][53][54][55][19,46,53,54,55].
Figure 3. Scheme showing the proposed mechanism for the biosynthesis of cyclotide kalata B1. The cyclization step in cyclotides is mediated by an asparaginyl endopeptidase (AEP)-like ligase. The cyclization and cleavage of the C-terminal pro-peptide from the cyclotide precursor protein happen at the same time through a transpeptidation reaction, involving an acyl-transfer step from the acyl-ligase intermediate to the N-terminal residue of the cyclotide domain [50]. The protease responsible for the N-terminal cleavage required for the cyclization has been identified as a papain-like protease [56][58]. As shown in the scheme, the kalata B1 protein precursor contains an ER signal peptide, an N-terminal pro-region, the N-terminal repeat (NTR), the mature cyclotide domain, and a C-terminal flanking region (tail, also known as CTR). Figure adapted from reference [24].
Asparaginyl endopeptidase (AEP)-like ligases have been shown to mediate the C-terminal cleavage and backbone-cyclization of the linear cyclotide precursor (Figure 3) [57][58][56,57], while papain-like cysteine proteases have also been found to participate in the N-terminal cleavage required for the AEP-mediated backbone cyclization [56][58]. Several AEP-like ligases have been shown to work in vitro, being able to cyclize different linear peptides, including cyclotide precursors polypeptides containing D-amino acids [57][59][60][61][62][56,59,60,61,62]. Protein-disulfide isomerases (PDIs) have also been shown to play an important role in the oxidative folding of cyclotides in vivo [63]. These findings open the exciting possibility of genetically engineered modified organisms for the bioproduction of cyclotides [64].
4. Chemical Synthesis
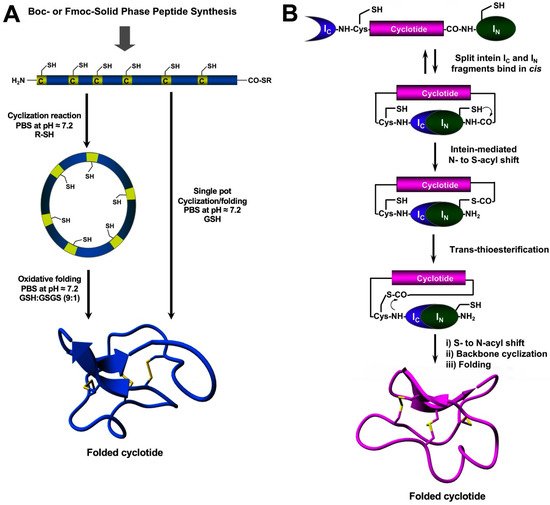
The chemical synthesis of cyclotides can be readily achieved using standard SPPS methods (for a detailed review on this topic, see [27]). Linear precursors can be easily produced by SPPS using Fmoc-based chemistry, the resulting linear precursors can be backbone-cyclized using an intramolecular version of native chemical ligation in aqueous buffers at physiological pH (pH ≈ 7) and then oxidatively folded sequentially (Figure 4A) [27]. A more convenient approach developed in theour lab involves performing the cyclization and folding steps in a ‘single pot’ reaction which requires only using reduced glutathione (GSH) as a thiol additive during the native chemical ligation reaction [65]. Using this approach, rwesearchers h have generated many disulfide-contained backbone-cyclized polypeptides [66][67][66,67], including naturally occurring and engineered cyclotides [5][33][35][66][5,33,35,66].
Figure 4. Different methods for the generation of native-folded cyclotides. (A) Chemical synthesis of cyclotides by employing an intramolecular version of native chemical ligation. This method requires the chemical production of a linear cyclotide precursor containing both an α-thioester moiety at the C-terminus and an N-terminal Cys residue. The linear precursor is then cyclized under reductive conditions and finally oxidatively folded using a proper redox buffer. The cyclization and oxidative folding reactions can be also efficiently performed in a ‘single pot’ reaction. This is accomplished by performing the cyclization in the presence of reduced GSH as the thiol cofactor. (B) Heterologous expression of cyclotides can be accomplished using protein trans-splicing (PTS) This approach has been employed for the generation of several MCoTI-cyclotides by using the native Cys residue located at the N-terminus of loop 6 to facilitate backbone cyclization. This method has been used to produce bioactive cyclotides using either eukaryotic or prokaryotic expression systems. Figure adapted from reference [23].
Chemically-produced cyclotide linear precursors have been also chemoenzymatically cyclized using purified AEP-like ligases [56][58][63][57,58,63]. This cyclization approach does not require the linear precursor to be natively folded [57][56]. A similar approach has also been used to produce trypsin inhibitor cyclotides from the corresponding linear precursors linearized at loop 1 but using trypsin [68]. In this case, the linear precursor linearized between the residues at the P1–P1′ junction requires to be natively folded to be recognized by the enzyme trypsin, which then facilitates the cyclization reaction [68]. The introduction of mutations that disrupt the binding between the linear precursor and the enzyme trypsin has been shown to reduce the cyclization yield [27]. The transpeptidase-like sortase A (SrtA) has been also employed for the backbone-cyclization of cyclotide precursors [69]. However, it should be mentioned that SrtA-mediated cyclizations require a recognition-specific sequence in the ligation site that is only partially removed during the transpeptidation reaction leaving an extra heptapeptide at the cyclization site.