2.Treatment and Repertoires治療とレパートリー
2.1. Changes in the Repertoire with ICIs
2.1. ICIによるレパートリーの変化
PD-1
blockade遮断により、主にエフェクター期の restores T cell functions primarily at the effector phase. Therefore, PD-1 blockade rescues exhausted CD8-positive T cells andT 細胞機能が回復します。したがって、PD-1 遮断は、使い果たされた CD8 陽性 T 細胞を救出し、その細胞毒性能力を回復させ、それによって腫瘍細胞の破壊を促進します [ restores50 their]。抗 cytotoxic capacity, thereby facilitating the destruction of tumor cells [10]. Anti-PD-1
antibodies prevent 抗体は、PD-1
from binding toが APC 上の PD-L1
on APCs and interfering with the differentiation and functioning of Treg cells because PD-1 plays a role in the development of Treg cellsに結合し、Treg 細胞の分化と機能を妨害するのを防ぎます。これは、PD-1 が Treg 細胞の発生に役割を果たすためです [11].[ However,51 the ]。ただし、PD-1
expression pattern encompasses a broad range of immune cells, including T cells, B cells, natural killer cells, dendritic cells, and bone marrow cells. Its ligands, の発現パターンには、T 細胞、B 細胞、ナチュラル キラー細胞、樹状細胞、骨髄細胞など、幅広い免疫細胞が含まれます。そのリガンドである PD-L1 (
programmed death-ligand 1) andプログラム死リガンド 1) と PD-L2
, are also は、がん細胞だけでなく、さまざまな造血細胞および非造血細胞でも発現しています [ expressed52、53 in]。したがって、抗 various hematopoietic and nonhematopoietic cells as well as cancer cells [12][13]. Therefore, anti-PD-1
therapy can affect various types of cells and pathways.療法は、さまざまな種類の細胞や経路に影響を与える可能性があります。
Recent analyses have mainly focused最近の分析は主に、腫瘍免疫応答で主要な役割を果たす on CD8-positive T cells, which play a major role in tumor immune responses. After anti-PD-1 therapy, most of the abundant TIL clones were found among T cell clonesCD8 陽性 T 細胞に焦点を当てています。抗 PD-1 治療後、豊富な TIL クローンのほとんどは、臨床反応に関係なく、末梢血中の T 細胞クローンに見られました [ in9 peripheral]。腫瘍抗原を認識する blood, regardless of the clinical response [9]. T
cell clones expressing TCR
s that recognize tumor antigens proliferate in anti-PD1 antibody を発現する T 細胞クローンは、抗 PD1 抗体応答者で増殖します[ 7、54 responders]。さらに、ネオアジュバント抗 [7][14]. Moreover, an analysis in patients who underwent neoadjuvant anti-PD-1
antibody抗体療法を受けた患者の分析では、メラノーマ特異抗原である therapy revealed a post-treatment increase in the number of CD8-positive T cells reactive with gp100, a melanoma-specific antigengp100 と反応する CD8 陽性 [15]. T
hese findings suggest細胞の数が治療後に増加することが明らかになった [ that55 ]。]。これらの知見は、使い果たされた抗腫瘍免疫応答の the ICI
-induced restoration of exhausted antitumor immune responses is followed by the clonal proliferation of immune cells, yielding therapeutic benefits. 誘導性の回復に続いて、免疫細胞のクローン増殖が起こり、治療上の利益をもたらすことを示唆しています。
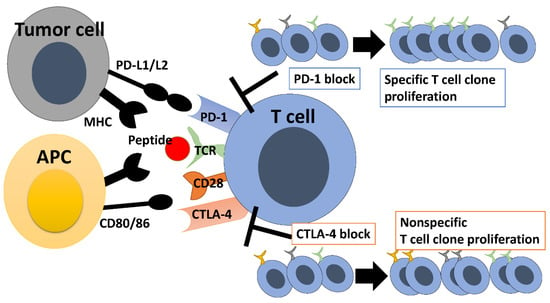
Meanwhile,一方、別の免疫チェックポイントタンパク質である blockade of CTLA-4
, another immune checkpoint protein, decreases the T cell priming の遮断は、T 細胞のプライミング閾値を低下させ、より多くのエフェクター T 細胞の増殖を可能にします [ threshold56 and]。抗 allows for the proliferation of more effector T cells [16]. Anti-CTLA-4
antibodies抗体は、メモリー also allow for the proliferation of memory T cell clonesT 細胞クローンの増殖も可能にします [ [17].57 Moreover,]。さらに、腫瘍微小環境の Treg
cells in the tumor microenvironment are depleted as they express 細胞は、CTLA-4
[18].を発現するため枯渇する Depletion[ of58 ]。Treg
cells improves intratumoral細胞の枯渇は、腫瘍内 IL-2
levels, facilitates survival of CD8-positive T cells, and expands the TCR repertoireレベルを改善し、CD8 陽性 T 細胞の生存を促進し、TCR レパートリーを拡大します [19].[ In59 other]。つまり、抗 words, anti-CTLA-4
antibodies inhibit Treg cells, reverse inhibit CD8-positive T cells, and expand the TCR repertoire in抗体は Treg 細胞を阻害し、逆に CD8 陽性 T 細胞を阻害し、非特異的に TCR レパートリーを拡大する [ a60]。さらに、抗 nonspecific manner [20]. In addition, anti-CTLA-4
antibodies accelerate the turnover of 抗体は、T
cell repertoire and increase TCR diversity細胞レパートリーの代謝回転を促進し、TCR の多様性を高めます[ [21][22][23].61、62、63 ]。抗 Anti-CTLA-4
antibodies broaden the T cell repertoire, whereas anti-PD1 antibodies promote the proliferation of a limited number of clones, skewing the T cell repertoire 抗体は T 細胞のレパートリーを広げますが、抗 PD1 抗体は限られた数のクローンの増殖を促進し、T 細胞のレパートリーをゆがめます (
Figure図 1 )
.
。
Figure図 1. Repertoire changes with immune checkpoint inhibitors.
免疫チェックポイント阻害剤によるレパートリーの変化。
A previous study showed tumor antigenic changes after PD-1 blockade
[24][64]. Thus, a dynamic clonal change in the TIL could be induced by antigenic alteration of tumor cells during treatment
[24][64], whereas the clone of TIL expanded after PD-1 blockade responded to tumor cell line before PD-1 blockade
[25][65]. Therefore, the authors reported that dynamic tumor-specific clonal changes after PD-1 blockade are caused by PD-1 blockade but not antigenic alteration of tumor cells
[25][65].
T cells use TCRs to recognize antigen peptides presented by MHC on antigen-presenting cells. CD28 receives stimulus from CD80/86, which leads to T cell activation. CTLA-4 competes with CD28 and inhibits T cell activation. Therefore, inhibition of CTLA-4 results in nonspecific proliferation of T cell clones and expansion of the TCR repertoire. On the other hand, T cells use TCRs to recognize antigen peptides on tumor cells for eliciting antitumor immune responses, and tumor PD-L1 binding to PD-1 on T cells inhibits the antitumor immune responses. PD-1 inhibition promotes the proliferation of specific T cell clones, leading to alteration of the TCR repertoire.
2.2. Changes in the Repertoire with BRAF/MEK Inhibition
MEK inhibitors can impair T cell activation, because T cell activation mediated by TCRs and their co-stimulatory molecules is dependent on mitogen-activated protein kinase (MAPK) and the PI3K-AKT signaling cascade
[26][66]. In fact, pharmacological in vitro inhibition of MEK had adverse effects on T cell activation
[27][67].
In contrast, the results of an in vivo analysis were twofold: MEK inhibition had no adverse effects on T cell effector function and showed favorable outcomes in combination with immune checkpoint inhibition
[28][68]. MEK inhibition was associated with increased tumor-infiltrating CD8-positive T cells, increased IFN-γ gene expression signatures, and decreased abundance of tumor-associated macrophages and Treg cells. Furthermore, MEK inhibition protected effector T cells from activation-induced cell death due to chronic TCR stimulation
[29][69]. In addition, an analysis in melanoma patients showed that BRAF/MEK inhibition upregulated the levels of T-bet and TCF7 and expanded T cell repertoire in tumors
[30][34]. Another study showed that higher intratumoral T cell clonality was associated with better responses to BRAF inhibitor treatment for melanoma
[31][70]. Based on these results, efficacy analyses of ICIs combined with BRAF/MEK inhibitors are desired in clinical trials.
2.3. Changes in the Repertoire with Adoptive Cell Transfer of TIL Therapy
Adoptive cell transfer (ACT) of TIL is used for the treatment of advanced melanoma. ACT of TIL has shown significant clinical benefit [
71]. However, ACT of TIL could not work enough in patients previously treated with PD-1 or MAPK inhibition [
68]. Response to ACT correlates with the recognition of tumor neo-antigens [
72,
73]. Anti-PD-1 naïve patients were received TIL reactive with more neo-antigens compared with anti-PD-1 experienced patients [
74]. Treatment products administered to anti-PD-1 naïve patients were more likely to contain T cells reactive against neoantigens than treatment products for anti-PD-1 experienced patients [
74].
2.4. Changes in the Repertoire with IL-12 Therapy
IL-12, an inflammatory cytokine, induces the proliferation and activation of natural killer (NK) cells and cytotoxic T cells and enhances effector functions
[32][33][75,76]. Additionally, it is an important link between innate and acquired immune systems, because APC-producing IL-12 stimulates the release of IFN-γ from T cells and NK cells
[34][77]. It is also involved in Th1 induction and promotes IFN-γ production
[35][78]. Thus, IL-12 plays a role in antitumor immunity, and T cells are important for IL-12-mediated tumor suppression
[36][79]. Intratumoral plasmid IL-12 electroporation therapy was tested in a phase II trial in melanoma patients
[37][80]. Following the treatment, intratumoral T cells proliferated clonally, which led to a skewed TCR repertoire.
2.5. Treatment Correlations and Repertoires
CTLA-4 is expressed mainly on CD4-positive T cells after TCR-mediated activation and interferes with CD28 co-stimulatory signaling induced by APCs to inhibit TCR-induced activation and proliferation
[38][81]. Therefore, CTLA-4 inhibition induces the activation and proliferation of antitumor T cells via increased CD28 signaling.
PD-1 inhibits the effector function of antigen-specific T cells upon binding to ligands
[39][82]. PD-1 inhibitors directly regulate the functions of various types of PD-1-expressing immune cells
[40][83]. Immunotherapy with anti-PD-1 antibodies is widely used for the treatment of metastatic solid tumors, with a response rate of 20–55%
[3]. Biomarkers to predict which patients are likely to respond to anti-PD-1 antibody therapy are needed.
Because highly variable CDR3 of the TCR chain is unique to individual T cell clones, CDR3 can be used to monitor the dynamics of T cell repertoire responses to ICIs
[41][84]. A recent study showed that the TCR clonality or diversity of T cells in the blood increases 3 weeks after the initiation of treatment with anti-PD-1 antibodies
[7]. Clonal proliferation of TCRs in the blood also occurred only in responders 3 weeks after the initiation of combination treatment with anti-PD-1 and anti-CTLA-4 antibodies. Thus, with this approach, minimally invasive liquid biopsies may be used in the early stages of treatment to predict patients’ treatment response.
The anti-PD-1 antibody treatment seems to be more effective in melanoma patients when the pretreatment TCR repertoire of TILs is larger in metastatic tumors
[42][85]. An analysis of melanoma in The Cancer Genome Atlas reveals that a larger TCR repertoire of TILs is associated with longer overall survival even without anti-PD-1 antibody treatment. Furthermore, TCR repertoires in the peripheral blood of melanoma patients were examined to determine whether the TCR diversity predicted the clinical prognosis of ICI treatment
[43][20]. Higher TCR repertoire diversity in the blood was associated with longer progression free survival, and low repertoire diversity was associated with poor prognosis. The diversity in patients who experienced late recurrence and long-term survival was significantly higher than that in rapid progressors. The TCR repertoire diversity in tumors may have a potential prognostic value.
Liquid biopsies were performed before treatment to predict responses to ICIs
[44][86]. In the PCR analysis of pretreatment peripheral blood mononuclear cells (PBMCs), the diversity evenness of the TCRs repertoire score was correlated with the therapeutic efficacy of ICIs. Furthermore, the pretreatment level of TCR repertoire restrictions in CD4-positive T cells in peripheral blood had a potential prognostic value for clinical response to CTLA4 inhibition
[43][20]. In addition, T cells release their DNA into the blood when cell death occurs. The released cell-free DNA in the blood was sequenced for analyzing CDR3 of T cells
[23][63], which suggested that the clonal proliferation of T cell repertoire in the blood within 3 weeks of starting ICI treatment predicts the therapeutic efficacy.
2.6. Immune-Related Adverse Events and Repertoires
Anti-CTLA-4 and anti-PD-1 therapies can prolong survival in melanoma patients; however, these therapies can also induce organ-specific toxicities, called immune-related adverse events (IrAEs), which make it impossible to continue ICIs in a considerable number of patients
[7][45][7,87]. It is likely that T cell clonal analysis is useful for the early diagnosis of IrAEs. The number of T cell clones that newly underwent proliferation was higher in patients with severe IrAEs after CTLA-4 blockade
[46][88]. Furthermore, newly expanded clones were found among CD8-positive T cells, but not CD4-positive T cells, in patients with IrAEs
[47][89]. In addition, severe repertoire restrictions were found in CD4-positive T cells in a study using samples of severe colitis
[48][22]. CD4-positive and CD8-positive T cells may be diversely involved in various IrAEs; further studies are necessary for clarifying this point.
2.7. Repertoire Analysis and Vaccines
Peptides recognized by TCRs are used as vaccines to enhance antitumor immune responses
[49][90]. Specifically, tumor biopsy specimens and nonmalignant tissue samples (usually PBMCs) are collected from patients and subjected to whole exome sequencing for comparison between tumor DNA and germline DNA to identify tumor-specific somatic mutations. A computational approach is used to predict MHC class I-binding epitopes, and peptides predicted to have moderate-to-high MHC-binding affinities are likely to induce CD8-positive T cell responses
[50][91].
MHC class II-binding peptides are often more difficult to predict. The peptide-binding groove of MHC class I has closed ends to define the arrangement of a peptide epitope composed of 8–11 amino acid residues for presentation to CD8-positive T cells. In contrast, the peptide-binding groove of MHC class II has open ends and can bind to a peptide that is longer and more variable in length. Recently, new methods have been developed for the prediction of MHC class II-binding peptides
[51][52][53][92,93,94], and they are expected to promote the development of MHC class II-binding peptides to elicit tumor-specific responses of CD4-positive T cells.