Neuroinflammation has been correlated with the progress of neurodegeneration in many neuropathologies. Although glial cells have traditionally been considered to be protective, the concept of them as neurotoxic cells has recently emerged. Thus, a major unsolved question is the exact role of astroglia and microglia in neurodegenerative disorders. On the other hand, it is well known that glucocorticoids are the first choice to regulate inflammation and, consequently, neuroglial inflammatory activity. The objective of this study was to determine how chronic dexamethasone treatment influences the host immune response and to characterize the beneficial or detrimental role of glial cells. To date, this has not been examined using a natural neurodegenerative model of scrapie. With this aim, immunohistochemical expression of glial markers, prion protein accumulation, histopathological lesions and clinical evolution were compared with those in a control group. Although impact of dexamethasone administration on neuropathological lesions was not demonstrated and treatment did not seem to be clinically relevant to disease progress when clinical signs had already begun, the evident extension of survival in one case was hopeful. The findings presented in this study support a potential failure of astrocytes and a stimulation of phagocytosis of PrPsc deposits by microglia. Thus, it is evidenced here how the complex interaction between glial populations failed to compensate for brain damage in natural conditions, emphasizing the need for using natural models. Additionally, the data showed that modulation of neuroinflammation by anti-inflammatory drugs might become a research focus as a potential therapeutic target for prion diseases, similar to that considered previously for other neurodegenerative disorders classified as prion-like diseases.
- dexamethasone
- neuroinflammation
- astrocytes
- microglia
- prion diseases
- neurodegeneration
Results
Clinical Signs
1. Clinical Signs
Clinical sheep, regardless of whether they were treated with DEX or not, showed motor symptoms (tremors, ataxia, incoordination and prostration) likely associated with deep affectation of the cerebellum, as well as pruritus in most cases. Table 1 shows the main clinical signs developed by each clinical sheep during the experiment. Control sheep did not present any of the key features of scrapie throughout the time of the experiment in any cases.
Table 1. Main clinical signs related to scrapie developed by both dexamethasone (DEX)-treated and non-treated clinical sheep.
| Group | Sheep No | Main Clinical Signs |
|---|---|---|
| Clinical non-treated | 11 | Tremors, pruritus, ataxia, lost look |
| 12 | Pruritus with skin lesions, alopecia, hyperexcitation | |
| 13 | Pruritus, alopecia, prostration | |
| 14 | Tremors, alopecia, prostration | |
| 15 | Pruritus | |
| Clinical DEX-treated | 16 | Pruritus, ataxia, tremors |
| 17 | Pruritus, alopecia | |
| 18 | Tremors, ataxia, hyperexcitation | |
| 19 | Pruritus, alopecia, hyperexcitation | |
| 20 | Tremors, intense widespread alopecia, prostration | |
| 21 | Hyperexcitation, prostration | |
| 22 | Tremors, constant pruritus, bruxism | |
| 23 | Scarce pruritus | |
| 24 | Intense pruritus with skin lesions | |
| 25 | Pruritus, alopecia, prostration |
As expected, DEX-treated sheep (mainly from the control group, due to the extended duration of treatment) experienced lesions compatible with secondary effects of the GC, including Cushing’s syndrome, in 3 out of 4 cases. Nevertheless, wound lesions were the major safety concern due to the long-term use of DEX (3 out of 4 control-treated sheep were affected by extensive alopecia).
Regarding survival time, although no statistically significant differences were observed between Kaplan ¬–Meier curves in clinical sheep, one of the ten (10%) treated clinical sheep survived 155 days compared to 72 days as the maximum observed for the non-treated clinical animals (Figure 1).
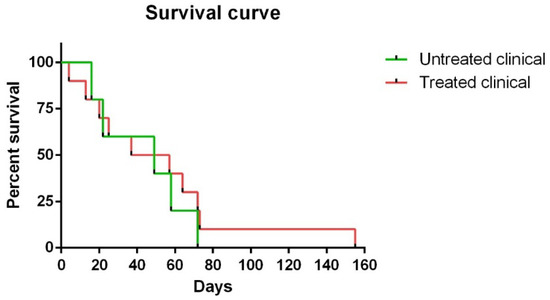
Figure 1. Kaplan–Meier survival curves corresponding to non-treated and treated clinical sheep. Note that one of ten treated clinical sheep survived 155 days, which was much longer than the maximum (72 days) observed for non-treated clinical animals.
Histopathological Findings (H-E)
2. Histopathological Findings (H-E)
Spongiosis was absent in all regions examined in sheep samples of both treated and non-treated control groups, while it was widespread in all areas in the clinical groups. Vacuolation was mainly located in the neuropil in all cases where it was found, although intraneuronal vacuoles were also observed in some cases. In regard to brain regions, subtle spongiform changes were found in the Fc, while they were much more pronounced and severe in the MO of all clinical animals. Thus, caudal tissues were the most affected by spongiform changes (Figure 2A). Regarding the specific impact of DEX administration on spongiform changes, no significant differences were found between treated and non-treated clinical sheep (p > 0.05) (Figure 2B).
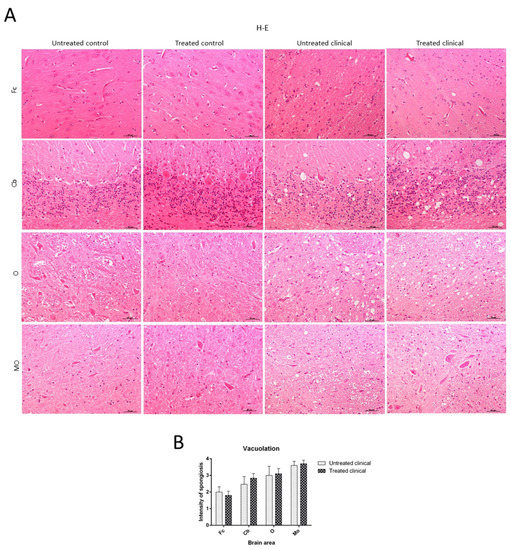
Figure 2. Vacuolation intensity (H-E staining). (A) Spongiosis was absent in all regions examined in both treated and non-treated control groups, while it was widespread in all tissues in clinical groups. Vacuolation was most frequently located in the neuropil. Note that spongiform change was most pronounced and severe in most caudal tissues. (B) Statistical analysis evidenced no differences between treated and non-treated clinical sheep. Medulla oblongata (MO), obex (O), cerebellum (Cb) and frontal cortex (Fc).
The significant decrease in motor activity observed in treated and non-treated clinical sheep was consistent with cell damage observed in the Purkinje cell layer, which presented severe vacuolation and even partial disappearance of Purkinje cells in some cases. Furthermore, these cells appeared swollen with neurite thickening (torpedoes) (Figure 3).
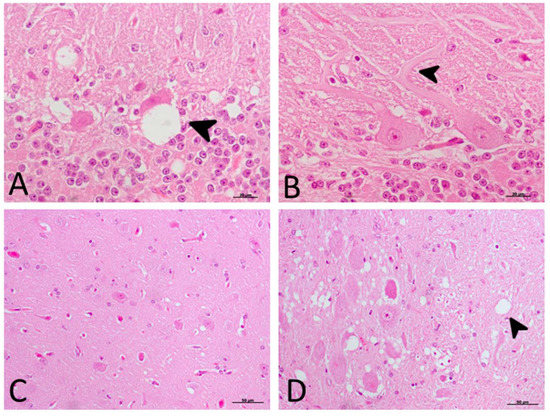
Figure 3. Main morphological findings in clinical sheep (H-E staining). (A) Cell damage observed in the Purkinje cell layer of cerebellum. Note the severe intraneuronal vacuolation (arrowhead) and partial disappearance of Purkinje neurons. (B) Purkinje cells appeared swollen with higher neurite thickening (torpedoes, arrowhead). (C) Subtle spongiform change was found in the frontal cortex. (D) Meanwhile, neuropil vacuolation (arrowhead) was pronounced and severe in the medulla oblongata in all cases.
Immunohistochemical (IHC) Findings
3. Immunohistochemical (IHC) Findings
PrP
3.1. PrP
sc
Accumulation
No PrPsc deposits were observed in any tissues belonging to control animals. On the other hand, prion protein deposition was widespread in all brain areas examined in both treated and non-treated clinical groups. Quantitative differences concerning PrPsc deposits were found between the Cb and Fc in both groups of clinical animals (Figure 4A). No significant differences between treated and non-treated clinical animals were found by the Mann–Whitney U test (Figure 4B).
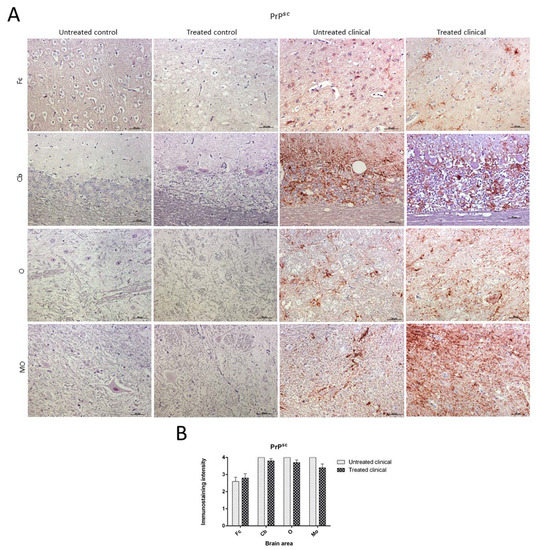
Figure 4. Pathological prion protein deposition by immunohistochemistry (IHC) with L42. (A) No PrPsc deposits were observed in any tissues belonging to animals from control groups. PrPsc deposition was widespread in all brain regions examined in both treated and non-treated clinical groups. (B) No statistical differences between treated and non-treated clinical sheep were found. Medulla oblongata (MO), obex (O), cerebellum (Cb) and frontal cortex (Fc).
Although lineal, spot, coalescent or granular PrPsc deposition patterns could be observed on some occasions, the coalescent pattern was the most frequently observed pattern in all brain regions. Purkinje cells in the cerebellum were never immunostained for this marker.
GFAP
3.2. GFAP
As previously described, paraffin sections processed for IHC showed an increase in GFAP immunoreactivity in clinical scrapie when compared with controls (Figure 5A).
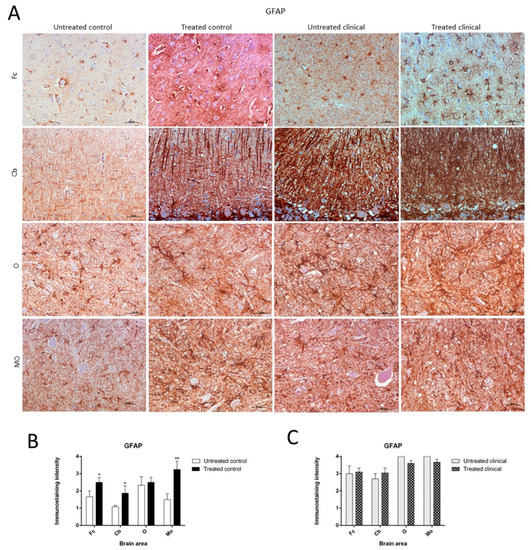
Figure 5. Glial fibrillary acidic protein (GFAP) immunostaining. (A) Increase of GFAP immunoreactivity in non-treated clinical scrapie compared with non-treated controls was evidenced. (B) Treated control animals showed significantly higher immunolabeling for GFAP than non-treated animals in all regions, except the obex. (C) GFAP immunostaining in the four regions examined did not reveal differences between treated and non-treated clinical animals. Medulla oblongata (MO), obex (O), cerebellum (Cb) and frontal cortex (Fc).
Treated control animals exhibited significantly higher immunolabeling for GFAP than the non-treated control group in all regions, except the obex (Fc, p < 0.05; Cb, p < 0.05; MO, p < 0.01) (Figure 5B). Strikingly, by contrast, GFAP immunostaining did not reveal major differences regarding the effect of treatment in any brain area examined in both clinical groups (Figure 5C). A multivariate linear regression analysis verified that the effect of treatment was significant (p < 0.05).
Morphologically, GFAP immunolabeling was found surrounding the meningeal zones in all regions examined. In the cerebellum, as previously described, the Purkinje cell layer was often immunostained, and an intense radial profile of GFAP in the molecular layer was found in samples with the highest intensity. Meanwhile, samples with the lowest GFAP intensity predominantly presented a horizontal profile in this layer of the cerebellum. In addition, the vast majority of astrocytes in the medulla oblongata and obex showed hypertrophic morphology in treated controls compared to astrocytes in the non-treated sheep, which demonstrated their typical stellate morphology (Figure 6).
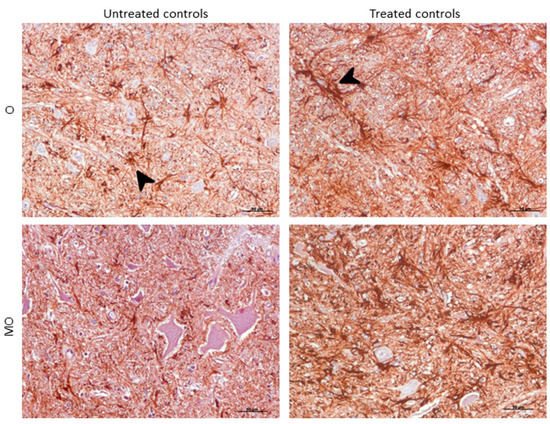
Figure 6. Morphological findings by GFAP IHC. Astrocytes in the medulla oblongata (MO) and obex (O) were widespread hypertrophic (arrowhead) after DEX treatment in controls compared to astrocytes in the non-treated ones, which appeared to be ramified (arrowhead).
IBA-1
3.3. IBA-1
As expected, the IHC technique demonstrated an expansion of microglial populations (IBA-1+ cells) in clinical animals compared with controls. However, no differences were found regarding the intensity of microgliosis among brain regions either for the control or clinical group (Figure 7A).
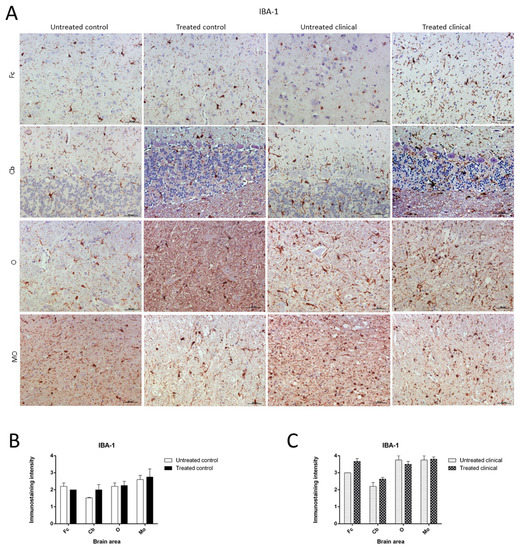
Figure 7. Ionized calcium-binding adaptor molecule-1 (IBA-1) immunostaining. (A) Microglial immunostaining intensity did not change with DEX treatment in the control and clinical groups. (B,C) No statistically significant differences in IBA-1 intensity between treated and non-treated animals were found. Medulla oblongata (MO), obex (O), cerebellum (Cb) and frontal cortex (Fc).
Contrary to those observed changes for astroglia in controls, the microglia immunostaining pattern did not significantly change upon DEX treatment during clinical or control stages. Thus, no statistically significant differences in intensity were found between microglia in treated and non-treated animals (Figure 7B,C).
Morphologically, the Purkinje cell layer in the cerebellum was never immunostained for IBA-1, and the ramified phenotype was the most frequently observed morphology. Additionally, a relevant higher percentage of microglial cells in this encephalic area presented an amoeboid phenotype in treated controls compared to microglia in non-treated sheep (Figure 8).
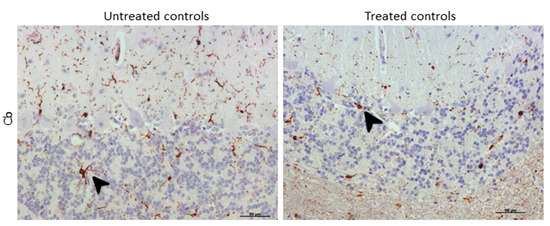
Figure 8. Morphological findings by IBA-1 IHC. A higher proportion of microglial cells in cerebellum presented an amoeboid (arrowhead) phenotype in DEX-treated controls compared to that in non-treated ones, which appeared to be mostly ramified (arrowhead).
RT-qPCR
3.4. RT-qPCR
Although it was not statistically significant (p = 0.064), daily injections of DEX in clinical sheep tended to show increased GFAP mRNA levels in the cerebellum compared with non-treated clinical animals. However, no significant differences were found in the mRNA expression of the microglial markers tested (AIF-1 and CD68 genes) in the cerebellum (Figure 9A).
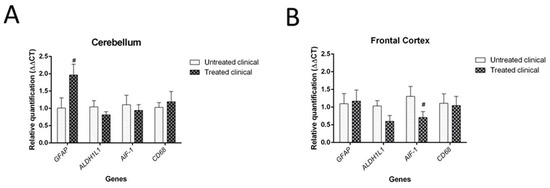
Figure 9. Results of gene expression by RT-qPCR in clinical individuals. (A) DEX treatment evidenced a tendency (#) toward increased GFAP mRNA levels in the cerebellum of treated animals compared with non-treated animals, although it did not reach statistical significance (p = 0.064). (B) Although it was not statistically significant (p = 0.085), there was a tendency toward decreased AIF-1 gene expression (microglia) in the frontal cortex of treated animals compared with that of animals in the non-treated group.
Regarding the frontal cortex, AIF-1 (or IBA-1) expression tended to be lower in clinical DEX-treated sheep compared with the non-treated group, although this was not statistically significant (p = 0.085). No changes were detected for astrocytic markers (GFAP and ALDH1L1) in this brain region (Figure 9B).
4. Discussion
Currently, no treatments for prion diseases exist. To our knowledge, most of the few studies testing potential treatments in scrapie as an archetype of this group of disorders were carried out many years ago. Moreover, their main focus was on targeting PrPsc or PrPc instead of the process of neuroinflammation. Here, the effect of long-term GC administration on survival period, PrPsc deposition, spongiosis and glial response was evaluated using naturally infected scrapie sheep as a natural model of neurodegenerative diseases. Thus, the purpose of this experiment was to determine how the synthetic GC DEX influences the host immune response in order to characterize the beneficial or detrimental role of glial cells in the progress of neurodegeneration of prion diseases. Previous studies testing different strategies to manipulate microglial function reported contrary effects on survival period in experimental models of prion diseases [1][2]. This prompted us to more deeply investigate the role of host immunity during the course of natural infection.
Intensive research efforts have been made to look for a therapeutic solution for neurodegenerative diseases based on immunomodulation. The development of therapeutic strategies that inhibit NF-kB activity was proposed to tackle these pathologies [3]. In fact, DEX had been used as a candidate glial cell modulator [4] and was found to protect neurons via decreased neuroinflammation of glial cells in a mouse model of PD [5]. More recently, GC therapy demonstrated clinical benefit in AD [6] and was associated with a lower risk of dementia in humans [7]. In contrast, GCs were described as neurotoxic for AD in murine models since they enhanced the pathology, augmenting deposits of amyloid beta and tau [8]. In regard to prion diseases, Outram et al., (1974) observed a reduction in the susceptibility to murine scrapie infection of mice treated with GCs [9]. Treatment with ibuprofen in murine scrapie was not conclusive due to the risky side effects [10].
The results described here regarding scrapie at clinical stages of disease partially agree with previous reports showing that no treatment is effective when neuronal degeneration has already begun. This is consistent with descriptions for non-steroidal anti-inflammatory drugs (NSAIDs) in AD [11], suggesting that prevention strategies are necessary instead of treatments. No beneficial long-term effect on disease progression was demonstrated for GC therapy in multiple sclerosis (MS) [12]. However, it is worth mentioning that, in the present study, one clinical sheep extended its survival period after DEX treatment. Even though this was only observed for one individual, it represents an encouraging result, as it opens up the possibility for anti-inflammatory therapy to have potential in at least some cases. Further studies to understand this potentiality are being designed.
Of note, corticosteroids have been established to cause dose-related immunosuppression, yet the mechanisms behind this impaired immune function have not been defined [13]. GCs exhibit paradoxical immunomodulatory functions [14][15]; although traditionally known as anti-inflammatory drugs, sometimes the modulating mechanisms fail, and they can aggravate CNS inflammation [16]. The paradoxical effects of GCs on neuronal survival and death have been attributed to the concentration and the ratio of receptor activation. GC-induced leucine zipper (GILZ) is a recently identified protein transcriptionally upregulated by GCs. Constitutively expressed in many tissues including the brain, GILZ mediates many of the actions of GCs. It mimics the anti-inflammatory and antiproliferative effects of GCs, but also exerts differential effects on stem cell differentiation and lineage development. Together with the dosage of GCs, the length of treatment in relation to the immune response is decisive to determine if GCs exhibit pro- or anti-inflammatory properties [17]. Acquired resistance is another problem, according to descriptions in MS after treatment for 3–6 months [18]. All these reasons could explain the lack of differences observed in this study regarding the length of treatment, despite varying from some weeks to nearly 18 months.
Although some studies argue that DEX has minimal access to the CNS [19][20][21], the present study clearly demonstrates the successful efficacy of DEX to cross the blood–brain barrier, as evidenced by the strong astroglial reaction in treated controls. However, this experiment also presented further complications due to the issue of GC therapy itself. DEX has a number of adverse effects, mostly associated with suppression of immunity [22]. The same side effects as those described by other authors [23][24] were observed in the present study, with outstanding wound loss as the most frequent and highly adverse side effect. On many occasions, animals needed to be euthanized because of this effect. For this reason, it was difficult to conclude whether this immunosuppressive therapy might ameliorate clinical signs or slow down the neurodegenerative process. The data provided herein should be interpreted with caution due to the use of a natural model, along with the inherent difficulties described above.
Regarding the specific impact of DEX administration on neuropathological lesions, no significant differences in vacuolation or PrPsc deposition were found between treated and non-treated clinical sheep. The observed cell damage in the Purkinje cell layer of the cerebellum is in agreement with previous findings in scrapie [25], Creutzfeldt–Jakob disease (CJD) [26] and other neurodegenerative disorders [27]. When these cells were closely observed in ultrastructural studies, the vacuolation occurring around this cell type displayed a close relationship with glial cells [28]. In regard to differences among brain regions, the caudal areas were the most affected by spongiform changes and PrPsc deposits. They were more frequent and widespread in the cerebellum compared to the frontal cortex, as previously reported in scrapie [29]. PrPsc presented different deposit patterns, although the coalescent pattern was most frequently observed in all brain regions, as was previously found in the cerebellum from CJD-affected individuals [26]. In contrast, a recent study demonstrated that PrPsc accumulates in all brain regions independently of neurodegeneration [30]; this discrepancy in results might be due to differences in models, since the study used a mouse experimental model of Gerstmann–Sträusler–Scheinker syndrome, and the other researchers used natural models of scrapie and CJD. This reinforces that we should be cautious with extrapolation from experimental models to reality.
Focusing on glial cells, which are the key topics in this study, both activated astrocytes and microglia, based on morphology as well as specific marker expression, were observed in clinical animals compared to controls. The activation of glia in scrapie [25][31] and other prion-like diseases [32][33] has been exhaustively characterized. However, this has not previously been described as a result of anti-inflammatory treatment.
In this study, a statistically significant difference in GFAP marker expression was observed in all regions examined, except the obex, after DEX treatment compared to controls. By contrast, treatment did not reveal major differences between clinical animals. This may be due to downregulation of astrogliosis, reflecting astroglial paralysis in the clinical stage of scrapie, as was recently described in late stages of AD [34][35]. Further studies would be essential to confirm this assertion.
The cerebellum is a preferential target of prions in scrapie [36][37] and CJD [38][39][40]. In fact, a relevant finding in this encephalic area could help to clarify astrocytic behavior in the neurodegenerative progress of prion diseases. An intense radial profile was observed in the molecular layer with high intensity, while the horizontal profile was instead related to samples with low intensity of this marker. This is the same as that evidenced in our previous work for human prion and prion-like disease samples [26][32]. As claimed then, this pattern suggested a possible glial stem cell response in order to protect against or compensate for neuronal loss [41]. This would agree with the hypothesis about astroglial paralysis, which, despite trying to react against brain damage, might prevent competent astrocytes from being formed. In the same vein, RT-qPCR results demonstrated a tendency toward increased GFAP mRNA, which is consistent with this assumption. Even though astroglia seem to attempt to compensate for the damage by initiating proliferation/regeneration, an error somewhere in this process could be blocking the final aim.
Another GFAP morphological finding was that the vast majority of astrocytes presented a hypertrophic morphology in treated control samples (similar to that at the clinical stage) in comparison with the typical stellate form in non-treated samples. It is well known that microglia undergo complex metamorphoses when they are reactive. However, an enigma of control of astroglial morphology in brain physiology is beginning to emerge [42]. Indeed, this finding is, to our knowledge, the first description of this phenomenon in astroglia.
In this in vivo study, comparing non-treated clinical and control samples, there was also an expansion of the microglial population in the clinical stage, as expected. This resulted in an increased number of microglia associated with an activated and phagocytic phenotype, as previously reported [43][44]. Nevertheless, microglial immunostaining intensity did not significantly change after DEX treatment (neither in the clinical nor control group). Similarly, a previous study [45] did not observe a decrease in microglial activation after lipopolysaccharide (LPS) induction of neuroinflammation and intranasal DEX treatment in mice, suggesting that the dose was not sufficient to reduce IBA-1 expression. However, other studies managed to reduce microglia activation with DEX sodium phosphate by using cell cultures [46]. The discrepancies in results may be due to different experimental models, pharmacological presentation or routes of DEX administration. Indeed, recent studies have shown that the effects of GCs on brain inflammatory responses are truly complex [47].
Concerning morphological findings, a high percentage of microglial cells in the cerebellum presented an amoeboid phenotype in treated controls compared to non-treated ones, which appeared to be more ramified. Provided that the amoeboid phenotype represents the most activated microglial shape [48][49][50] associated with the expression of neuroinflammatory genes [51] and the presence of this amoeboid phenotype in prion diseases seems to be stimulated by the high accumulation of PrPsc [52], we might speculate that DEX treatment stimulates phagocytosis of PrPsc deposits, which would constitute a useful tool against prion progress. However, this stimulation was not evident in clinical animals despite the same treatment administration. This is consistent with the idea of a failure of the glial response.
Regarding gene expression analysis, DEX treatment in clinical sheep involved an increase in GFAP and a decrease in AIF-1 (also known as IBA-1) mRNA expression in the cerebellum and frontal cortex, respectively. This finding could be related to the previously described region-specific pattern of neuroinflammation in sporadic CJD [53], in accordance with different cytokine profiles found in these two brain regions. It is worth mentioning that glia immunostaining and its respective mRNA expression were not correlated in recently reported previous studies [45][54], an aspect that needs further clarification. Nevertheless, in this study, the tendencies demonstrated by high-resolution molecular analysis confirmed the overall results provided by IHC analysis. Consequently, both techniques, which herein were demonstrated to complement one another, led to the conclusion of a probable glial failure.
It is currently assumed that glial responses can play both protector and toxic roles depending on the degree of activation [55][56][57], supporting the concept that neuroinflammation induced by glia can amplify pathology [58][59]. The transition from neuroprotective to neurotoxic activity of astrocytes by cytokine stimulation had been demonstrated [4][60][61], but such neurotoxicity was prevented when astrocytes were treated with DEX in cell culture, while DEX had no effect on neurons. On the basis of these observations, DEX could promote neuroprotective properties of astrocytes, as confirmed here in the natural model of prion disease. Nevertheless, the findings presented in this study support a potential failure of astrocytes and not a role for enhancing pathology.
A recent study reported that neurotoxic astrocytes (called A1) are potential contributors to neuronal death in several neurodegenerative disorders [62][63]. This subtype of astrocytes might be involved in damaging actions, although the interaction of both populations, astroglia and microglia, has been postulated as a candidate involved in neurodegeneration [63][64], resulting in the essential presence of microglia [64]. As a consequence, drugs to block the release of neurotoxins by these astrocytes have been suggested as a possible solution [63]. Currently, the activation of astrocytes remains poorly understood. The release of cytokines from those astrocytes and microglia accompanies this event. Provided that the present study was focused on glial cells, the next goal would consist of providing new information about the mechanisms underlying cytokine release. As has been previously suggested, it could be a crucial target for therapeutic approaches in CNS in prion diseases [61]. It is indispensable to study how glial communication might prevent neuronal damage.
Distinct conformations of PrPsc might explain the unusual wide range of neuropathological, biochemical and clinical features of prion diseases [65] and may contribute to this disagreement in conclusions regarding treatments. Since a natural model was used in this study, it is likely that natural Transmissible Spongiform Encephalopathies (TSE) infections of ruminants involve mixtures of strains rather than a single strain [66], but it is necessary to emphasize that it would reflect the reality much better.
In conclusion, we would like to reassert the essential requirement of using natural models to provide reliable results. It constitutes a powerful in vivo approach to assess the activation of glial cells by anti-inflammatory therapy in a trustworthy and quantitative manner. However, it is also much more time consuming and entails inherent difficulties and uncontrollable factors, mainly the exact time of natural infection in the field. Transgenic models have demonstrated poor representativeness of natural disease progress; some examples include studies that sought to delay the progress of amyotrophic lateral sclerosis (ALS) in a murine model [67]. Unfortunately, anti-inflammatory treatment worsened symptoms in clinic phase III in ALS-affected humans [68] or in epidemiological studies with drugs that managed to reduce the risk of developing PD and AD [69], while clinical assays in sick patients failed [70].
Taking into consideration the overall results presented in this study and that some authors postulated that anti-inflammatory therapeutic approaches could be combined with other strategies to achieve improved therapeutic effects [71], combining this strategy with a natural model, such as that described in the present study, may confer an appropriate starting point to advance the subject.
References
- Diego Gomez-Nicola; Nina L. Fransen; Stefano Suzzi; V. Hugh Perry; Regulation of Microglial Proliferation during Chronic Neurodegeneration. The Journal of Neuroscience 2013, 33, 2481-2493, 10.1523/JNEUROSCI.4440-12.2013.
- James A. Carroll; Brent Race; Katie Williams; James Striebel; Bruce Chesebro; Microglia Are Critical in Host Defense against Prion Disease. Journal of Virology 2018, 92, e00549-18, 10.1128/jvi.00549-18.
- Mariagrazia Grilli; M Memo; Nuclear factor-kappaB/Rel proteins: a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochemical Pharmacology 1999, 57, 1–7.
- Liudmila Efremova; Petra Chovancova; Martina Adam; Simon Gutbier; Stefan Schildknecht; Marcel Leist; Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Archives of Toxicology 2017, 91, 231-246, 10.1007/s00204-016-1702-2.
- Iwona Kurkowska-Jastrzebska; Tomasz Litwin; Ilona Joniec-Maciejak; Agnieszka Ciesielska; Adam Przybyłkowski; A. Czlonkowski; Anna Czlonkowska; Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson's disease. International Immunopharmacology 2004, 4, 1307-1318, 10.1016/j.intimp.2004.05.006.
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056.
- Michal Schnaider Beeri; James Schmeidler; Gerson T. Lesser; Maria Maroukian; Rebecca West; Stephanie Leung; Michael Wysocki; Daniel P. Perl; Dushyant P. Purohit; Vahram Haroutunian; et al. Corticosteroids, but not NSAIDs, are associated with less Alzheimer neuropathology. Neurobiology of Aging 2012, 33, 1258-1264, 10.1016/j.neurobiolaging.2011.02.011.
- Michael Nerius; Britta Haenisch; Willy Gomm; Gabriele Doblhammer; Anja Schneider; Glucocorticoid Therapy is Associated with a Lower Risk of Dementia. Journal of Alzheimer's Disease 2020, 73, 175-183, 10.3233/jad-190444.
- G. W. Outram; A. G. Dickinson; H. Fraser; Reduced susceptibility to scrapie in mice after steroid administration. Nature 1974, 249, 855-856, 10.1038/249855a0.
- Constanze Riemer; Michael Burwinkel; Anja Schwarz; Sandra Gültner; Simon W. F. Mok; Ines Heise; Nikola Holtkamp; Michael Baier; Evaluation of drugs for treatment of prion infections of the central nervous system. Journal of General Virology 2008, 89, 594-597, 10.1099/vir.0.83281-0.
- Bruno Pietro Imbimbo; An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer's disease. Expert Opinion on Investigational Drugs 2009, 18, 1147-1168, 10.1517/13543780903066780.
- Denise Tischner; Holger M. Reichardt; Glucocorticoids in the control of neuroinflammation. Molecular and Cellular Endocrinology 2007, 275, 62-70, 10.1016/j.mce.2007.03.007.
- Le Min; F. Stephen Hodi; Ursula B. Kaiser; Corticosteroids and immune checkpoint blockade. Aging 2015, 7, 521-522, 10.18632/aging.100797.
- Allan Munck; Anikó Náray-Fejes-Tóth; The ups and downs of glucocorticoid physiology Permissive and suppressive effects revisited. Molecular and Cellular Endocrinology 1992, 90, C1-C4, 10.1016/0303-7207(92)90091-j.
- L. P. Filaretova; T. Podvigina; T. Bagaeva; O. Morozova; Dual action of glucocorticoid hormones on the gastric mucosa: how the gastroprotective action can be transformed to the ulcerogenic one. Inflammopharmacology 2009, 17, 15-22, 10.1007/s10787-008-8046-3.
- Shawn Sorrells; Javier R. Caso; Carolina D. Munhoz; Robert M. Sapolsky; The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33-39, 10.1016/j.neuron.2009.09.032.
- Spies, C.M.; Strehl, C.; van der Goes, M.C.; Bijlsma, J.W.; Buttgereit, F; Glucocorticoids. Best Pract Res. Clin. Rheumatol. 2011, 25, 891–900.
- John R. Kirwan; Glucocorticoid resistance in patients with rheumatoid arthritis. Scandinavian Journal of Rheumatology 2007, 36, 165-166, 10.1080/03009740701340263.
- Edo R. De Kloet; Brain Corticosteroid Receptor Balance in Health and Disease. Endocrine Reviews 1998, 19, 269-301, 10.1210/er.19.3.269.
- O. C. Meijer; Penetration of Dexamethasone into Brain Glucocorticoid Targets Is Enhanced in mdr1A P-Glycoprotein Knockout Mice. Endocrinology 1998, 139, 1789-1793, 10.1210/en.139.4.1789.
- Cara Hueston; Terrence Deak; The inflamed axis: The interaction between stress, hormones, and the expression of inflammatory-related genes within key structures comprising the hypothalamic–pituitary–adrenal axis. Physiology & Behavior 2014, 124, 77-91, 10.1016/j.physbeh.2013.10.035.
- Amber J. Giles; Marsha-Kay N. D. Hutchinson; Heather Sonnemann; Jinkyu Jung; Peter E. Fecci; Nivedita M. Ratnam; Wei Zhang; Hua Song; Rolanda Bailey; Dionne Davis; Caitlin M. Reid; Deric M. Park; Mark R. Gilbert; Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. Journal for ImmunoTherapy of Cancer 2018, 6, 51, 10.1186/s40425-018-0371-5.
- Günther Weindl; M. Schaller; Monika Schäfer-Korting; H.C. Korting; Hyaluronic Acid in the Treatment and Prevention of Skin Diseases: Molecular Biological, Pharmaceutical and Clinical Aspects. Skin Pharmacology and Physiology 2004, 17, 207-213, 10.1159/000080213.
- Michael W. Whitehouse; Anti-inflammatory glucocorticoid drugs: reflections after 60 years. Inflammopharmacology 2011, 19, 1-19, 10.1007/s10787-010-0056-2.
- Rodrigo S. Hernández; Rocío Sarasa; Adolfo Toledano; Juan J. Badiola; Marta Monzón; Morphological approach to assess the involvement of astrocytes in prion propagation. Cell and Tissue Research 2014, 358, 57-63, 10.1007/s00441-014-1928-3.
- Monzón, M.; Hernández, R.S.; Garcés, M.; Sarasa, R.; Badiola, J.J. Glial alterations in human prion diseases, a correlative study of astroglia, reactive microglia, protein deposition, and neuropathological lesions. Medicine (Baltimore) 2018, 97, e0320.
- Justyna R Sarna; Richard Hawkes; Patterned Purkinje cell death in the cerebellum. Progress in Neurobiology 2003, 70, 473-507, 10.1016/s0301-0082(03)00114-x.
- Rocío Sarasa; Concepción Junquera; Adolfo Toledano; Juan José Badiola; Marta Monzón; Ultrastructural changes in the progress of natural Scrapie regardless fixation protocol. Histochemistry and Cell Biology 2015, 144, 77-85, 10.1007/s00418-015-1314-6.
- Stephane Lezmi; T. Seuberlich; Anna Oevermann; T. Baron; Anna Bencsik; Comparison of Brain PrPd Distribution in Ovine BSE and Scrapie. Veterinary Pathology 2011, 48, 1101-1108, 10.1177/0300985810395784.
- James Alibhai; Richard A. Blanco; Marcelo A. Barria; Pedro Piccardo; Byron Caughey; V. Hugh Perry; Tom C Freeman; Jean Manson; Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration. PLoS Biology 2016, 14, e1002579, 10.1371/journal.pbio.1002579.
- Rocío Sarasa; Alfredo Martinez; Eva Monleón; Rosa Bolea; Antonia Vargas; Juan José Badiola; Marta Monzón; Involvement of astrocytes in transmissible spongiform encephalopathies: a confocal microscopy study. Cell and Tissue Research 2012, 350, 127-134, 10.1007/s00441-012-1461-1.
- Moisés Garcés; M. Isabel Guijarro; Antonia Vargas; Juan J. Badiola; Marta Monzón; Neuroglial patterns are shared by cerebella from prion and prion-like disorder affected patients.. Mechanisms of Ageing and Development 2019, 184, 111176, 10.1016/j.mad.2019.111176.
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al.et al Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421.
- Verkhratsky, A.; Marutle, A.; Rodriguez-Arellano, J.J.; Nordberg, A. Glial Asthenia and Functional Paralysis, A New Perspective on Neurodegeneration and Alzheimer’s Disease. Neuroscientist 2015, 21, 552–568.
- Alexei Verkhratsky; Jose Julio Rodrigues; Augustas Pivoriunas; Robert Zorec; Alexey Semyanov; Astroglial atrophy in Alzheimer's disease.. Pflüger, Archiv für die Gesammte Physiologie des Menschen und der Thiere 2019, 471, 1247-1261, 10.1007/s00424-019-02310-2.
- A. Williams; P.J. Lucassen; D. Ritchie; M. Bruce; PrP Deposition, Microglial Activation, and Neuronal Apoptosis in Murine Scrapie. Experimental Neurology 1997, 144, 433-438, 10.1006/exnr.1997.6424.
- H. Fraser; K. L. Brown; K. Stewart; I. McConnell; P. McBride; A. Williams; Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. Journal of General Virology 1996, 77, 1935-1940, 10.1099/0022-1317-77-8-1935.
- J Berciano; M T Berciano; J M Polo; J Figols; J Ciudad; Vanesa Lafarga; Creutzfeldt-Jakob disease with severe involvement of cerebral white matter and cerebellum.. Virchows Arch. Pathol. Anat. Histopathol. 1990, 417, 36–45.
- Armstrong, R.A.; Ironside, J.W.; Lantos, P.L.; Cairns, N.J. A quantitative study of the pathological changes in the cerebellum in 15 cases of variant Creutzfeldt-Jakob disease (vCJD). Neuropathol. Appl. Neurobiol. 2009, 35, 36–45.
- Ignazio Cali; Cathleen J. Miller; Joseph E. Parisi; Michael D. Geschwind; Pierluigi Gambetti; Lawrence B. Schonberger; Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone. Acta Neuropathologica Communications 2015, 3, 37, 10.1186/s40478-015-0214-2.
- María-Isabel Álvarez; Luis Rivas; César Lacruz; Adolfo Toledano Gasca; Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with Alzheimer's disease: A histological and immunohistochemical comparison. Glia 2015, 63, 287-312, 10.1002/glia.22751.
- Zeug, A.; Muller, F.E.; Anders, S.; Herde, M.K.; Minge, D.; Ponimaskin, E.; Henneberger, C; Control of astrocyte morphology by Rho GTPases. Brain Res. Bull. 2018, 126, 44–53.
- Hugh V. Perry; Colm Cunningham; Delphine Boche; Atypical inflammation in the central nervous system in prion disease. Current Opinion in Neurology 2002, 15, 349-354, 10.1097/00019052-200206000-00020.
- V. Hugh Perry; Vincent O'connor; The Role of Microglia in Synaptic Stripping and Synaptic Degeneration: A Revised Perspective. ASN Neuro 2010, 2, 281-291, 10.1042/AN20100024.
- G. Meneses; G. Gevorkian; A. Florentino; M. A. Bautista; A. Espinosa; G. Acero; G. Díaz; A. Fleury; I. N. Pérez Osorio; A. Del Rey; G. Fragoso; Edda Sciutto; H. Besedovsky; Intranasal delivery of dexamethasone efficiently controls LPS‐induced murine neuroinflammation. Clinical & Experimental Immunology 2017, 190, 304-314, 10.1111/cei.13018.
- Hui, B.; Yao, X.; Zhang, L.; Zhou, Q. Dexamethasone sodium phosphate attenuates lipopolysaccharide-induced neuroinflammation in microglia BV2 cells. Naunyn Schmiedebergs Arch. Pharmacol. 2020.
- Duque Ede, A.; Munhoz, C.D; The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol (Lausanne) 2016, 7, 78.
- Richard M. Ransohoff; V. Hugh Perry; Microglial Physiology: Unique Stimuli, Specialized Responses. Annual Review of Immunology 2009, 27, 119-145, 10.1146/annurev.immunol.021908.132528.
- Delphine Boche; V. H. Perry; J. A. R. Nicoll; Review: Activation patterns of microglia and their identification in the human brain. Neuropathology and Applied Neurobiology 2013, 39, 3-18, 10.1111/nan.12011.
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I; Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142.
- Kim, S.U.; de Vellis, J; Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313.
- H. Mühleisen; J. Gehrmann; R. Meyermann; Reactive microgIia in Creutzfeldt-Jakob disease. Neuropathology and Applied Neurobiology 1995, 21, 505-517, 10.1111/j.1365-2990.1995.tb01097.x.
- Franc Llorens; Irene Lã³Pez-Gonzã¡lez; Katrin Thã¼Ne; Margarita Carmona; Saima Zafar; Olivier Andrã©Oletti; Inga Zerr; Isidre Ferrer; Irene López-González; Katrin Thüne; et al.Olivier Andréoletti Subtype and Regional-Specific Neuroinflammation in Sporadic Creutzfeldt–Jakob Disease. Frontiers in Aging Neuroscience 2014, 6, 198, 10.3389/fnagi.2014.00198.
- Diana M. Norden; Paige J. Trojanowski; Emmanuel Villanueva; Elisa Navarro; Jonathan P. Godbout; Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300-16, 10.1002/glia.22930.
- Carroll, J.A.; Chesebro, B; Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65.
- DiSabato, D.J.; Quan, N.; Godbout, J.P; Neuroinflammation, the devil is in the details. J. Neurochem. 2016, 139, 136–153.
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D; The Role of Microglia in Prion Diseases, A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207.
- Liberski, P.P.; Sklodowski, P.; Klimek, A; Creutzfeldt-Jakob disease, ultrastructural study of brain biopsy, unusual interaction between astrocytes and oligo- and microglia. Folia. Neuropathol. 1995, 33, 85–92.
- Kordek, R.; Nerurkar, V.R.; Liberski, P.P.; Isaacson, S.; Yanagihara, R.; Gajdusek, D.C; Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9754–9758.
- Buffo, A.; Rolando, C.; Ceruti, S; Astrocytes in the damaged brain, molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89.
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W; Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167.
- Shane A. Liddelow; Ben A. Barres; Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957-967, 10.1016/j.immuni.2017.06.006.
- Shane A. Liddelow; Kevin A. Guttenplan; Laura E. Clarke; F. Chris Bennett; Christopher J. Bohlen; Lucas Schirmer; Mariko L. Bennett; Alexandra E. Münch; Won-Suk Chung; Todd C. Peterson; et al.Daniel K. WiltonArnaud FrouinBrooke A. NapierNikhil PanickerManoj KumarMarion S. BuckwalterDavid H. RowitchValina L. DawsonTed M. DawsonBeth StevensBen A. Barres Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481-487, 10.1038/nature21029.
- Kelly S. Kirkley; Katriana A. Popichak; Maryam F. Afzali; Marie E. Legare; Ronald B. Tjalkens; Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. Journal of Neuroinflammation 2017, 14, 99, 10.1186/s12974-017-0871-0.
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernandez-Borges, N.; Vanni, I.; Vaccari, G.; Marin-Moreno, A.; Frassanito, P.; et al.et al PrPC Governs Susceptibility to Prion Strains in Bank Vole, While Other Host Factors Modulate Strain Features. J. Virol. 2016, 90, 10660–10669.
- Candace K. Mathiason; Scrapie, CWD, and Transmissible Mink Encephalopathy. Progress in Molecular Biology and Translational Science 2017, 150, 267-292, 10.1016/bs.pmbts.2017.07.009.
- Jasna Kriz; Minh Dang Nguyen; Jean-Pierre Julien; Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis.. Neurobiology of Disease 2002, 10, 268-278, 10.1006/nbdi.2002.0487.
- Paul Gordon; Dan H Moore; Robert G Miller; Julaine M Florence; Joseph L Verheijde; Carolyn Doorish; Joan F Hilton; G Mark Spitalny; Robert B MacArthur; Hiroshi Mitsumoto; et al.Hans E NevilleKevin BoylanTahseen MozaffarJerry M BelshJohn RavitsRichard S BedlackMichael C GravesLeo F McCluskeyRichard J BarohnRup Tandan Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. The Lancet Neurology 2007, 6, 1045-1053, 10.1016/s1474-4422(07)70270-3.
- Ji-Kyung Choi; Bruce G. Jenkins; Isabel Carreras; Sukru Kaymakcalan; Kerry Cormier; Neil W. Kowall; Alpaslan Dedeoglu; Anti-inflammatory treatment in AD mice protects against neuronal pathology. Experimental Neurology 2010, 223, 377-384, 10.1016/j.expneurol.2009.07.032.
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J; et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression, a randomized controlled trial. JAMA 2003, 289, 2819–2826.
- Michael Burwinkel; Constanze Riemer; Anja Schwarz; Julia Schultz; Sabine Neidhold; Theresa Bamme; Michael Baier; Role of cytokines and chemokines in prion infections of the central nervous system. International Journal of Developmental Neuroscience 2004, 22, 497-505, 10.1016/j.ijdevneu.2004.07.017.