Heterogeneous catalysis is an important research hotspot in the global field. Due to its economic and energy-saving features, it is widely used in the transformation of various energy resources (e.g., petroleum, coal, natural gas, and solar energy) in nature, the synthesis of numerous industrial chemicals, the purification of vehicle exhaust, and so on. It is estimated that catalysts are used in the production processes of about 80% of artificial chemicals at some stages, and about 35% of the world’s gross domestic product (GDP) is accounted for by catalytic processes. In the field of catalysis, exploring the efficient, stable, and economical catalyst formulations has promoted the rapid development of nanomaterials synthesis methods. Traditional heterogeneous catalysts are metal nanoparticles (NPs) dispersed on supports. Among them, noble metals are the most efficient catalysts.
- single-atom catalysts
- SACs
- maximum atomic utilization
- catalytic activity
1. Introduction
2. CO Oxidation
3. Methane Combustion
4. Volatile Organic Compounds (VOCs) Oxidation
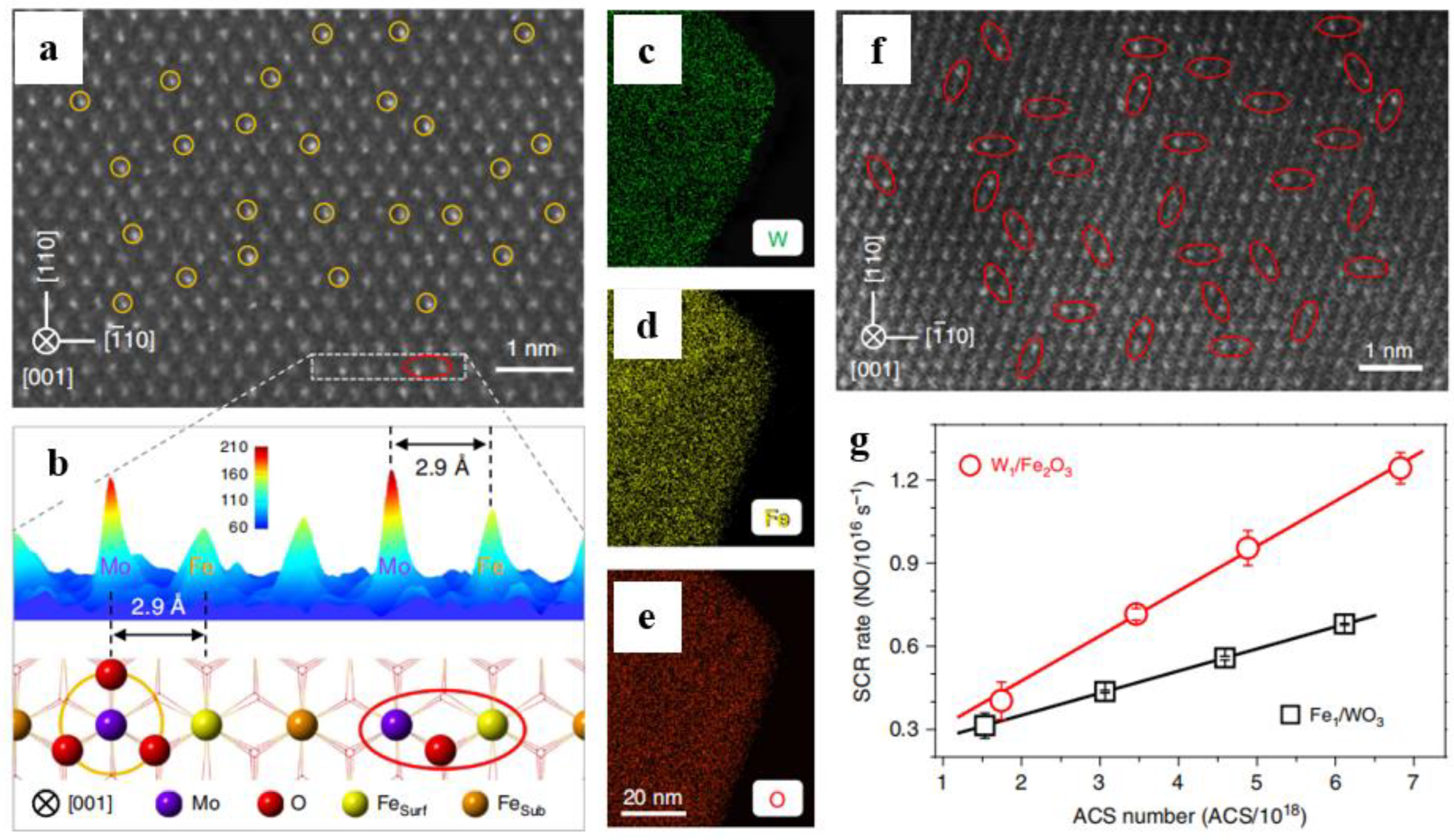
5. NOx Reduction
References
- Heiz, U.; Sanchez, A.; Abbet, S.; Schneider, W.D. Catalytic oxidation of carbon monoxide on monodispersed platinum clusters: Each atom counts. J. Am. Chem. Soc. 1999, 121, 3214–3217.
- Qiao, B.T.; Wang, A.Q.; Yang, X.F.; Allard, L.F.; Jiang, Z.; Cui, Y.T.; Liu, J.Y.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634.
- Wang, C.L.; Gu, X.K.; Yan, H.; Lin, Y.; Li, J.J.; Liu, D.D.; Li, W.X.; Lu, J.L. Water-mediated Mars−van Krevelen mechanism for CO oxidation on ceria-supported single-atom Pt1 catalyst. ACS Catal. 2017, 7, 887–891.
- Wang, H.; Shen, J.H.; Huang, J.F.; Xu, T.J.; Zhu, J.R.; Zhu, Y.H.; Li, C.Z. Atomically dispersed Au catalysts supported on CeO2 foam: Controllable synthesis and CO oxidation reaction mechanism. Nanoscale 2017, 9, 16817–16825.
- Luo, L.L.; Chen, S.Y.; Xu, Q.; He, Y.; Dong, Z.J.; Zhang, L.F.; Zhu, J.F.; Du, Y.G.; Yang, B.; Wang, C.M. Dynamic atom clusters on AuCu nanoparticle surface during CO oxidation. J. Am. Chem. Soc. 2020, 142, 4022–4027.
- Yoo, M.; Yu, Y.S.; Ha, H.; Lee, S.; Choi, J.S.; Oh, S.; Kang, E.; Choi, H.; An, H.; Lee, K.S.; et al. A tailored oxide interface creates dense Pt single-atom catalysts with high catalytic activity. Energy Environ. Sci. 2020, 13, 1231–1239.
- Qiao, B.T.; Lin, J.; Wang, A.Q.; Chen, Y.; Zhang, T.; Liu, J.Y. Highly active Au1/Co3O4 single-atom catalyst for CO oxidation at room temperature. Chin. J. Catal. 2015, 36, 1505–1511.
- Zhang, Z.L.; Zhu, Y.H.; Asakura, H.; Zhang, B.; Zhang, J.G.; Zhou, M.X.; Han, Y.; Tanaka, T.; Wang, A.Q.; Zhang, T.; et al. Thermally stable single atom Pt/m-Al2O3 for selective hydrogenation and CO oxidation. Nat. Commun. 2017, 8, 16100.
- Lou, Y.; Cai, Y.F.; Hu, W.D.; Wang, L.; Dai, Q.G.; Zhan, W.C.; Guo, Y.L.; Hu, P.; Cao, X.M.; Liu, J.Y.; et al. Identification of active area as active center for CO oxidation over single Au atom catalyst. ACS Catal. 2020, 10, 6094–6101.
- Ding, K.L.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189.
- Pereira-Hernández, X.I.; DeLaRiva, A.; Muravev, V.; Kunwar, D.; Xiong, H.F.; Sudduth, B.; Engelhard, M.; Kovarik, L.; Hensen, E.J.M.; Wang, Y.; et al. Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen. Nat. Commun. 2019, 10, 1358.
- Wang, H.; Liu, J.X.; Allard, L.F.; Lee, S.; Liu, J.L.; Li, H.; Wang, J.Q.; Wang, J.; Oh, S.H.; Li, W.; et al. Surpassing the single-atom catalytic activity limit through paired Pt−O−Pt ensemble built from isolated Pt1 atoms. Nat. Commun. 2019, 10, 3808.
- Maurer, F.; Jelic, J.; Wang, J.; Gänzler, A.; Dolcet, P.; Wöll, C.; Wang, Y.; Studt, F.; Casapu, M.; Grunwaldt, J.D. Tracking the formation, fate and consequence for catalytic activity of Pt single sites on CeO2. Nat. Catal. 2020, 3, 824–833.
- Guo, L.W.; Du, P.P.; Fu, X.P.; Ma, C.; Zeng, J.; Si, R.; Huang, Y.Y.; Jia, C.J.; Zhang, Y.W.; Yan, C.H. Contributions of distinct gold species to catalytic reactivity for carbon monoxide oxidation. Nat. Commun. 2016, 7, 13481.
- Kropp, T.; Mavrikakis, M. Transition metal atoms embedded in graphene: How nitrogen doping increases CO oxidation sctivity. ACS Catal. 2019, 9, 6864–6868.
- Qiao, B.T.; Liu, J.X.; Wang, Y.G.; Lin, Q.Q.; Liu, X.Y.; Wang, A.Q.; Li, J.; Zhang, T.; Liu, J.Y. Highly efficient catalysis of preferential oxidation of CO in H2-rich stream by gold single-atom catalysts. ACS Catal. 2015, 5, 6249–6254.
- Xiong, H.F.; Kunwar, D.; Jiang, D.; García-Vargas, C.E.; Li, H.Y.; Du, C.C.; Canning, G.; Pereira-Hernandez, X.I.; Wan, Q.; Lin, S.; et al. Engineering catalyst supports to stabilize PdOx two-dimensional rafts for water-tolerant methane oxidation. Nat. Catal. 2021, 4, 830–839.
- Hou, Z.Q.; Dai, L.Y.; Deng, J.G.; Zhao, G.F.; Jing, L.; Wang, Y.S.; Yu, X.H.; Gao, R.Y.; Tian, X.R.; Dai, H.X.; et al. Electronically engineering water resistance in methane combustion with an atomically dispersed tungsten on PdO catalyst. Angew. Chem. Int. Ed. 2022, 61, e202201655.
- Hackett, S.F.J.; Brydson, R.M.; Gass, M.H.; Harvey, I.; Newman, A.D.; Wilson, K.; Lee, A.F. High-activity, single-site mesoporous Pd/Al2O3 catalysts for selective aerobic oxidation of allylic alcohols. Angew. Chem. Int. Ed. 2007, 46, 8593–8596.
- Jiang, Z.Y.; Feng, X.B.; Deng, J.L.; He, C.; Douthwaite, M.; Yu, Y.K.; Liu, J.; Hao, Z.P.; Zhao, Z. Atomic-scale insights into the low-temperature oxidation of methanol over a single-atom Pt1-Co3O4 catalyst. Adv. Funct. Mater. 2019, 29, 1902041.
- Jiang, Z.Y.; Jing, M.Z.; Feng, X.B.; Xiong, J.C.; He, C.; Douthwaitee, M.; Zheng, L.R.; Song, W.Y.; Liu, J.; Qu, Z.G. Stabilizing platinum atoms on CeO2 oxygen vacancies by metal-support interaction induced interface distortion: Mechanism and application. Appl. Catal. B 2020, 278, 119304.
- Huang, Z.W.; Gu, X.; Cao, Q.Q.; Hu, P.P.; Hao, J.M.; Li, J.H.; Tang, X.F. Catalytically active single-atom sites fabricated from silver particles. Angew. Chem. Int. Ed. 2012, 51, 4198–4203.
- Zhang, C.B.; Liu, F.D.; Zhai, Y.P.; Ariga, H.; Yi, N.; Liu, Y.C.; Asakura, K.; Flytzani-Stephanopoulos, M.; He, H. Alkali-metal-promoted Pt/TiO2 opens a more efficient pathway to formaldehyde oxidation at ambient temperatures. Angew. Chem. Int. Ed. 2012, 51, 9628–9632.
- Chen, J.; Jiang, M.Z.; Xu, W.J.; Chen, J.; Hong, Z.X.; Jia, H.P. Incorporating Mn cation as anchor to atomically disperse Pt on TiO2 for low temperature removal of formaldehyde. Appl. Catal. B 2019, 259, 118013.
- Yang, K.; Liu, Y.X.; Deng, J.G.; Zhao, X.T.; Yang, J.; Han, Z.; Hou, Z.Q.; Dai, H.X. Three-dimensionally ordered mesoporous iron oxide-supported single-atom platinum: Highly active catalysts for benzene combustion. Appl. Catal. B 2019, 244, 650–659.
- Cui, X.J.; Xiao, J.P.; Wu, Y.H.; Du, P.P.; Si, R.; Yang, H.X.; Tian, H.F.; Li, J.Q.; Zhang, W.H.; Deng, D.H.; et al. A graphene composite material with single cobalt active sites: A highly efficient counter electrode for dye-sensitized solar cells. Angew. Chem. Int. Ed. 2016, 55, 6708–6712.
- Zhang, H.Y.; Sui, S.H.; Zheng, X.M.; Cao, R.R.; Zhang, P.Y. One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures. Appl. Catal. B 2019, 257, 117878.
- Fujiwara, K.; Pratsini, S.E. Single Pd atoms on TiO2 dominate photocatalytic NOx removal. Appl. Catal. B 2018, 226, 127–134.
- Lin, J.; Qiao, B.T.; Li, N.; Li, L.; Sun, X.C.; Liu, J.Y.; Wang, X.D.; Zhang, T. Little do more: A highly effective Pt1/FeOx single-atom catalyst for the reduction of NO by H2, Chem. Commun. 2015, 51, 7911–7914.
- Xie, S.H.; Tan, W.; Li, Y.J.; Ma, L.; Ehrlich, S.N.; Deng, J.G.; Xu, P.; Gao, F.; Dong, L.; Liu, F.D. Copper single atom-triggered niobia−ceria catalyst for efficient low-temperature reduction of nitrogen oxides. ACS Catal. 2022, 12, 2441–2453.
- Chen, J.X.; Fang, X.; Ren, Z.H.; Qu, W.Y.; Hu, X.L.; Ma, Z.; Chen, L.W.; Liu, X.; Chen, Y.X.; Tang, X.F. Single Mo atoms paired with neighboring Ti atoms catalytically decompose ammonium bisulfate formed in low-temperature SCR. J. Mater. Chem. A 2022, 10, 6065.
- Qu, W.Y.; Liu, X.N.; Chen, J.X.; Dong, Y.Y.; Tang, X.F.; Chen, Y.X. Single-atom catalysts reveal the dinuclear characteristic of active sites in NO selective reduction with NH3. Nat. Commun. 2020, 11, 1532.