Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Marta Morante and Version 2 by Sirius Huang.
Metastatic melanoma is a highly immunogenic tumor with very poor survival rates due to immune system escape-mechanisms. Immune checkpoint inhibitors (ICIs) targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and the programmed death-1 (PD1) receptors, are being used to impede immune evasion.
- melanoma
- inhibitors
- immunotherapy
1. Introduction
Melanoma is unquestionably the most aggressive form of skin cancer. It generally arises due to the accumulation of genetic mutations in melanocytes, the pigment-producing cells in the skin, mostly as a consequence of overexposure to sunlight. Once the disease has extended from the initial lesion to become metastatic, the prognosis is very poor and the final outcome is fatal in the majority of the cases. The American Cancer Society estimates that there will be more than 99,780 cases and 7,650 deaths in the United States caused by melanoma in 2022 [1]. With a historical perspective, annual melanoma incidence rates are escalating as rapidly as 4–6% [2].
Melanoma is characterized by being a highly immunogenic tumor [3]. Such a feature should induce adaptive immune responses in the organism, aimed at preventing tumor progression. For this, it is necessary that melanoma cells present adequate amounts of antigens, both qualitative and quantitatively, in order to trigger immune activation, instead of immune tolerance. This process depends on multiple factors pertaining both to the tumor cells themselves and the surrounding microenvironment [4]. In order to generate an effective anti-tumor immune response, seven steps, constituting what has been termed the Cancer–Immunity Cycle [5], must be enacted: (i) The release of cancer cell antigens; (ii) Cancer-specific antigen presentation by dendritic cells or antigen-presenting cells (APCs); (iii) Priming and activation of cytotoxic T-lymphocytes (CTLs) against the cancer-specific antigens that have been recognized as foreign; (iv) CTLs transportation to the tumor vicinity; (v) CTLs infiltration into the tumor; (vi) Recognition and binding to cancer cells by the CTLs; and (vii) The killing of the targeted cancer cells.
In spite of their potential immunogenicity, melanoma cells have developed mechanisms of immune escape, based on attenuating the response of the tumor microenvironment [6]. This can be achieved by: (i) impeding an optimal activation of melanoma-infiltrating lymphocytes [7][8][7,8]; (ii) through the inhibition of CTLs’ function, either by the up-regulation of immune checkpoint ligands [9] or by stimulating the populations of immune suppressive cells such as myeloid-derived suppressor cells (MDSCs) [10] or regulatory T lymphocytes (Tregs) [11]; (iii) or by evoking CTLs’ death by apoptosis. In addition, other pro-tumorigenic effects, such as the stimulation of tumor angiogenesis and stroma remodeling [12], facilitate melanoma cells’ avoidance of the immune response.
2. Use of Immune Checkpoints Inhibitors in the Treatment of Melanoma
The interactions among tumor cells, APCs and T cells, as well as among T cells and the rest of the body’s cells, is orchestrated by a plethora of stimulatory and inhibitory molecules that regulate T cell activation [13]. Stimulatory molecules play a key role in activating the immune system. These molecules can regulate T cells’ responses by amplifying signals, carried out by co-stimulatory receptors, or counteracting signals, orchestrated by the co-inhibitory receptors [14]. So, in order to be active, the T cell has to be activated by two signals. First, the T-cell receptor (TCR) must recognize a part of a specific foreign epitope. The second signal is delivered by a co-stimulatory molecule. Without this last signal, the T cell is not fully active and it becomes anergic or dies. Once the T-cell activation is not needed, the co-inhibitor receptors come into play and curtail T-cell activation. Throughout all this process, the organism has immunological checkpoints, based on the action of inhibitory molecules, which prevent unwanted and harmful self-directed activities that lead to autoimmunity [15].
Immune checkpoints inhibitors (ICIs) are attracting enormous attention for the treatment of metastatic melanoma. The co-inhibitory receptors CTLA4 (cytotoxic T-lymphocyte-associated protein 4) and PD1 (programmed death-1) are two of the aforementioned inhibitory molecules, whose activation slows down the activity of the CTLs, resulting in an attenuation of the immune response against tumor cells [15]. However, melanoma cells have learned to utilize these inhibitory devices to their advantage, thereby evading immune destruction. For instance, the expression of CTLA4 in T cells is upregulated in melanoma cells, which provides that the T cells cannot be fully activated [16]. Moreover, these cells can also elude immune surveillance by expressing PDL1 (programmed death-ligand 1), the PD1 ligand, which ends up suppressing the T-cell function [17]. As such, during the last decade, the use of antibodies has been directed against the immune checkpoints such as CTLA4 and PD1, impeding the union between B7-CTLA4 and PD1-PDL1, respectively, (Figure 1). ICIs have yielded impressive clinical benefits for the treatment of metastatic melanoma, which has led to their approval both by the US and European drug agencies [18]. These ICIs have the goal of blocking specific immune checkpoint molecules in order to enhance intrinsic anticancer immunity [19].
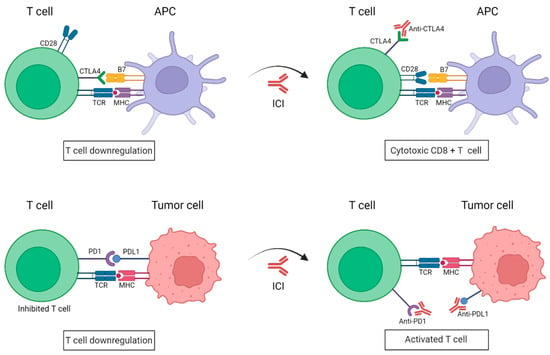
Figure 1. Schematic representation of immune checkpoint blockade by antibodies anti-PD1 and anti-CTLA4. Under normal conditions, the binding of PD1 expressed on activated T cells to its ligand PDL1 on tumor cells causes T-cell exhaustion. CTLA4 and CD28 are found in T cells and they compete with each other to bind their ligand B7 present on APCs. CTLA4 has a higher affinity to B7 than CD28 and transmits an inhibitory signal to T cells, whereas CD28 transmits a T-cell stimulatory signal. In the presence of ICIs, anti-PD1/PDL1 antibodies impede the union of PD1 to its ligand PDL1, resulting in a killed tumor cell; whereas anti-CTL4 antibodies work by blocking the inhibitory B7-CTLA-4 signaling, allowing T cells to be active. APC: Antigen-presenting cell; TCR: T-cell receptor; MHC: major histocompatibility complex; PD1: programmed death-1; PDL1: programmed death-ligand 1; CTLA4: cytotoxic T lymphocyte antigen. (Created with BioRender; www.biorender.com, access date: 20 October 2022).
CTLA4 is expressed on CD8+ and CD4+ T cells. Ipilimumab (anti-CTLA4 monocolonal antibody) was the first ICI to be approved by the FDA for the treatment of metastatic melanoma [20]. It is capable of preventing the CTLA4-induced CTL inhibition, which ends up with T-cell activation. Similarly, nivolumab and pembrolizumab target the interaction between PD1 with its ligands PDL1 (also known as B7-H1 or CD274) and PDL2 (also known as B7-DC or CD273) and have also obtained FDA approvals for the treatment of patients with unresectable or metastatic melanoma [21]. PD1 is found in the surface of T and B lymphocytes, natural killer cells and some myeloid populations. Upon its blockade by an antibody, its immunomodulatory function will be impaired, which will allow the T cells to continue and be active against the tumor [22]. Other antibodies against the ligand PDL1, expressed in the tumoral cells, are atezolizumab and durvalumab, which are also being tested in different clinical trials for the treatment of melanoma. Recently, a new clinical trial is incorporating a new anti-CTLA4 antibody named quavonlimab [23].
Before the appearance of antibodies against ICIs, patients with metastatic melanoma had a 5-year survival rate of 23% [24]. The therapeutic options consisted of antineoplastic chemotherapy drugs (such as dacarbazine) and high doses of interleukin-2 (IL-2), which resulted in severe adverse effects and low, overall survival (OS) benefit. With the implementation of ICIs, alone or in combination, the survival figures improved, with a rise in OS to 50% and response rates around 40% [25][26][25,26]. Immunotherapy provides long-lasting responses (more than 30 months) in almost one third of the patients. However, immune-related adverse events (irAEs) are often associated with such treatments. These include tissue-specific inflammatory responses, more often appearing associated with therapies against CTLA4 than to those targeting PD1 [27]. The most common ailments include pruritus, rash, nausea, diarrhea and thyroid disorders [28], but most of them are manageable. Contrarily, clinical trials have unveiled that the combination of anti-CTL4 and anti-PD1 therapeutics, despite exhibiting improved clinical benefits, displays a significant surge in irAEs grade 3 or 4 which, in some cases, calls for the discontinuation of the treatment [29][30][29,30].
Unfortunately, initial ICI-responder patients develop disease progression in a period of time. A clinical trial to test Ipilimumab efficacy in unresectable stage III or IV metastatic melanoma showed that 60% of patients maintained the response for at least 2 years [31]. However, the disease progressed in non-responder patients or patients that acquired resistance mechanisms. Similarly, a high percentage of patients treated with the PD1/PDL1 inhibitor, pembrolizumab, lose response over the time [32][33][32,33]. Immunotherapy-acquired resistance mechanisms are due to the evasion of immune recognition of tumoral cells. Among others, intratumor heterogeneity and low neo-antigens presentation in tumor cells [34][35][34,35], exclusion of T cells from the tumor microenvironment [36][37][38][36,37,38] and modulation of T-cell function in an immunosuppressive tumor microenvironment [39][40][39,40], such as presence of myeloid cells or low levels of oxygen, seem to be the cause of such evasion [41]. Presence of myeloid-derived suppressor cells contribute to CD8+ T-cell apoptosis [39], that can be avoid by blocking the Fas/Fas ligand pathway, therefore enhancing the immunotherapy efficiency [42]. Moreover, T-cell apoptosis can be triggered by the tumor antigen CD73, whose nucleotidase activity contributes to the tumoral immune evasion [40].
The molecular mechanism underlying resistance to ICIs remains elusive and limited, pointing to dysregulated cancer metabolism and epigenetic alterations as the drivers of immunotherapy escape. Recent study of the disease evolution over 9 years of a metastatic melanoma patient have shown some insights into the acquired resistance mechanisms [43][44][43,44]. Most of the tumor samples exhibited PTEN loss and genome duplication causing instability and aneuploidy. Moreover, few driver-alterations were identified, such as CDKN2A, epigenetic alterations and DNA-damage sensors’ dysregulation. However, cancer drivers such as BRAF or H/N/KRAS oncogenes were not found. The burden of immune cells in the tumor microenvironment was also diminished in resistant metastasis. Together with other studies, the common features during immunotherapy resistance are B-catenin activation [44][45][46][44,45,46], PTEN loss [37][47][37,47], lack of response to IFNg [48][49][48,49], depletion of tumor-specific neo-antigen presentation [49][50][49,50], genome instability [51][52][51,52], cell-cycle dysregulation [53] and epigenetic modulations [38][54][38,54].