Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Aishwarya A Makam and Version 2 by Jessie Wu.
Type-1-diabetes (T1D) is a multifactorial disorder with a global incidence of about 8.4 million individuals in 2021. It is primarily classified as an autoimmune disorder, where the pancreatic β-cells are unable to secrete sufficient insulin. This leads to elevated blood glucose levels (hyperglycemia). The development of T1D is an intricate interplay between various risk factors, such as genetic, environmental, and cellular elements. Here, the focus is on cellular elements such as ER stress leading to defects in insulin secretion and β-cell destruction.
- ER stress
- T1D
- amyloids
- insulin granule
1. A Concise Overview of the Pancreas
Type-1-diabetes (T1D)D is linked with dysregulated hormone secretions from the pancreas [1][2][1,11]. The pancreas is a 15–20 cm, leaf shaped organ located in the upper abdomen, behind the stomach in humans. Histologically, it comprises both exocrine and endocrine tissues [2][11]. The bulk of the pancreatic tissue, consisting of acinar cells, performs the exocrine function. The exocrine functions include the production of digestive enzymes for protein, carbohydrate, and fat digestion. Masked in a nest of acinar cells are patches of the endocrine tissue, the Islets of Langerhans. Rightly referred to as micro-organs, close to one million of these exist and cover about 1–2% of the total pancreatic mass.
2. Endoplasmic Reticulum S stress and Its Consequences in Type-1-1Diabetes
Among all the membrane bound organelles of a eukaryotic cell, the endoplasmic reticulum (ER) is one of the most essential one. The ER is responsible for the proper folding of membrane and secretory proteins, synthesis of sterols and lipids, and a reserve for free Ca++ [3][32]. The lumen of the rough ER, which gets its title from the numerous ribosomes attached to its surface, provides an optimal microenvironment for protein processing [3][32]. Cells need optimal ER functioning to maintain homeostasis. The demand for functional proteins is higher in secretory cells, such as the islet β-cells. The β-cells have a basal level of translation required for cellular maintenance. In addition to this, they must process large amounts of hormones or enzymes rapidly in response to physiological cues [4][33]. This leads to an additional burden on the ER, resulting in ER stress. Apart from this, other external factors such as viral infection and exposure to toxins also result in ER stress. Other studies have shown that there is a close association between ER stress and ROS production. Overall, these factors cause a drop in the efficiency of ER function, leading to the accumulation of mutated, unfolded, or misfolded proteins [4][5][33,34]. ER stress could be characterized as a disequilibrium between the demand for functional proteins and the ability of the ER to fold the proteins [6][35]. Specialized signaling pathways are activated in response to ER stress. The most well-characterized facet of ER stress signaling is the unfolded protein response (UPR) [7][36]. ER stress engages the adaptive UPR pathway, which upregulates the production of ER chaperones and causes the phosphorylation of the α subunit of translation initiation factor 2 (eIF2α), to temporarily repress global translation [8][9][10][11][37,38,39,40]. If these stress response pathways are unable to restore proteostasis, which leads to activation of terminal UPR, which upregulates pro-apoptotic factors, such as CHOP/GADD153, and drives the cell to apoptosis [8][37]. Cell injury due to chronic ER stress is being increasingly recognized as a common contributor to diabetes [12][41].
The connection between ER stress and T1D pathogenesis was first drawn from the observation that ER stress markers were upregulated in T1D β-cells compared to healthy β-cells. A direct link for β-cell ER stress and T1D was then provided by showing that T1D pathogenesis could be halted by mitigating β-cell ER stress [13][42]. The major result of ER stress in β-cells are the hurdles in protein processing. One well-studied example is the islet amyloid polypeptide (IAPP or amylin), a peptide hormone co-secreted with insulin. Pro-islet amyloid polypeptide (proIAPP) is produced as a 67 amino acid pro-peptide and undergoes post-translational modifications, including protease cleavage in the Golgi complex and insulin secretory granules, to produce mature IAPP [14][15][16][43,44,45]. Mature IAPP is stored in the insulin secretory granule and is found in the halo region of the granule, while insulin is located in the dense core [16][45]. Physiological levels of IAPP are approximately 1% those of insulin levels and are co-secreted during insulin exocytosis [14][43]. The IAPP released during insulin exocytosis plays a role in glycemic regulation by slowing gastric emptying and promoting satiety. Thereby, it prevents postprandial spikes in blood glucose levels [17][46].
3. Islet Amyloid PolypeptidePP and Amyloids
Islet amyloid polypeptide (IAPP) is characterized by its unique ability to aggregate and form toxic, insoluble, and pro inflammatory amyloid aggregates [14][16][43,45]. These aggregates are present in the majority of individuals with T2D and have been widely studied in T2D pathophysiology [18][19][47,48]. The possible role of IAPP amyloids in T1D pathology remains relatively unexplored in large sample sets. A study from the Better Diabetes Diagnosis cohort revealed that ~11% of patients with T1D had markedly elevated plasma IAPP levels relative to C-peptide and pro-insulin levels [19][48]. The accumulation of IAPP amyloid aggregates in β-cells induces ER stress, triggering endoplasmic reticulum-associated protein degradation (ERAD) and an unfolded protein response (UPR), as discussed before.
This has also been investigated at the single cell level, using cell line models (INS1E, etc.) by overexpressing human IAPP (hIAPP), where ER stress causes IAPP cytotoxicity [20][49]. ER stress is artificially induced in these cell lines using chemicals such as thapsigargin. Glucose-stimulated insulin secretion of INS1E cells having higher IAPP levels was comparable to cells with physiological levels of IAPP [20][49]. However, induction of ER stress coupled with hIAPP overexpression at high glucose (16.7 mM) severely diminishes glucose-stimulated insulin release from hIAPP-INS1E cells compared with untreated control cells. Treatment with chemical chaperones that alleviate ER stress (measured using ER stress markers such as PERK, CHOP, etc.) can rescue this hIAPP-induced cytotoxicity [20][49]. Last year, it was found that TXNIP, a protein upregulated during ER stress, is a strong transcriptional activator of IAPP in islet cells [21][50]. Thus, ER stress may possibly trigger greater amyloid formation, by increasing IAPP levels. This may indicate a positive feedback loop, as depicted in Figure 1, that eventually induces apoptosis and autoimmune activity.
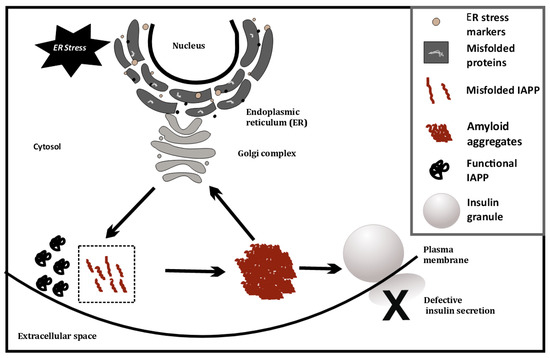
Figure 1.
ER stress-induced misfolding of IAPP in β-cells, leading to amyloid formation.
4. Effects of ER Stress on Large Dense Core Vesicles—Insulin Secretory Granules (ISGs)
Large dense core vesicles (LDCVs) are vesicles specialized for the storage of secretory proteins. LDCVs are formed in the trans-Golgi network (TGN), where specific pH and redox conditions prevail. The core of these vesicles is dense and contains large amounts of granulogenic protein aggregates. β-cells produce many such LDCVs for insulin storage and secretion. These are known as insulin secretory granules (ISGs) [22][23][65,66] (Figure 2). A fraction of these granules is trafficked to the plasma membrane to prepare them for release. An elaborate process of docking and priming precedes the fusion of these vesicles with the membrane [24][25][26][27][67,68,69,70], ultimately releasing insulin upon the increase in cytoplasmic Ca++ [28][71]. Many of the proteins involved in this process are similar to the ones involved in synaptic transmission [29][72]. More recently, proteins that are not found in synaptic transmission have been identified as unique players [24][29][67,72]. These have been shown to be involved in β-cell secretion. This underscores the importance of the further investigations required in this area. Some of these players have a huge relevance in ER stress and the development of T1D [30][31][58,73]. Proteins such as TSPAN7 and ZnT8, involved in trafficking of insulin granules, are also known to act as autoantigens during T1D. ZnT8 was first identified as a major T1D autoantigen by exploiting the bioinformatics approaches on β-cell-specific proteins [32][33][64,74]. The presence of ZnT8 autoantibodies has been further documented in 60–80% of Caucasian patients [32][64] with type-1-diabetes, and in 58% of patients in the Japanese population [34][75]. ZnT8 is a key member of the insulin secretory granules (ISGs) and is mainly responsible for the intake of zinc ions into the granules [34][75] (Figure 2). ZnT8 may also regulate the function of proinsulin processing enzymes such as Pro convertase 1/2 (PC1/2), which have zinc ions as a cofactor [32][64]. Zinc ions stabilize the stored form of insulin and mediate the release of insulin from secretory granules [33][34][74,75]. It is likely that recognition of ZnT8 as an autoantigen on ISG membranes has a negative impact on insulin secretion and processing during T1D (Figure 2). The binding of autoantibodies to ZnT8 may also affect its function as an ion transporter, leading to a deficiency of zinc in insulin secretory granules. It has previously been observed that knock outs of ZnT8 show a loss of the dense core of Zn-insulin crystals, which results in abnormalities in glucose tolerance, insulin processing, and secretion [33][35][74,76]. Impaired functioning of ZnT8, due to recognition as a T1D autoantigen, may thus promote β-cell dysfunction, insulin release, and an inflammatory microenvironment.
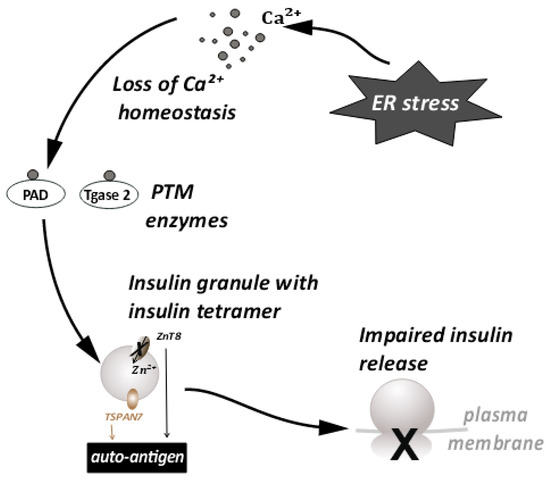
Figure 2. ER stress disturbs the Ca++ ion homeostasis. This leads to the conversion of proteins important for insulin granule processing and release into autoantigens. This further leads to impaired insulin release.
Tetraspanins are transmembrane molecules that act as protein organizers and play a role in vesicle trafficking in the neurons. Tetraspanin 7 expression was also detected in the pancreatic islets [36][77] (Figure 2). β-cells, being secretory in nature are enriched with L-type Ca++ channels. These Ca++ channels are important in the glucose stimulated insulin response (GSIS) [37][78]. Tetraspanin 7 has been shown to be involved in regulating the function of the L-type Ca++ channels of the β-cells [38][79]. In TSPAN7 knockdown (KD) cells, at a basal glucose concentration of 7 mM, a modest elevation in Ca++ concentration was observed. Insulin secretion is pulsatile [39][80] and it synchronizes with Ca++ oscillations. The frequency of these oscillations is increased in TSPAN7 KD cells. This disturbs the cellular homeostasis, since it is important for the β-cells to maintain a fine balance between insulin production and insulin secretion [22][65].
The production of insulin is regulated by a cis element in the 5′ UTR of the preproinsulin mRNA (ppIGE). This regulation occurs in a glucose-dependent manner [40][81]. Glucose stimulation enhances proinsulin translation tenfold [40][81]. This imposes a burden on the ER of the insulin-producing β-cells. Supportive of this view, it has also been shown that the reduction of insulin production alleviates ER stress [20][41][42][49,82,83]. Given that ER functioning is also needed for the processing and folding of proinsulin [41][43][82,84], ER stress results in altered forms of insulin that are autoantigenic [44][45][55,85]. The recognition of insulin as an autoantigen further leads to inflammation, cytokine release, and an autoimmune response, culminating in T1D [46][86].
5. Conclusion
ER stress is predominant in the β-cells of T1D patients. Major repercussions of ER stress are defects in protein processing and autoantigen generation. The scope of future research lies in investigating the specific trafficking proteins that are affected due to ER stress, so that the consequences for insulin release can be reversed. The specific autoantigens leading to inflammatory response can be countered, as possible treatment strategies for type-1 diabetics.