Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Aleksandra Anna Zasada and Version 2 by Conner Chen.
Only three Corynebacterium species are known to produce a lethal exotoxin called diphtheria toxin. These are C. diphtheriae, C. ulcerans and C. pseudotuberculosis.
- NTTB
- diphtheria toxin
- Corynebacterium
- tox gene
1. Introduction
The genus Corynebacterium was first described in 1896 as a Gram-positive club-shaped bacillus with filamentous morphology [1]. The genus Corynebacterium belongs to the Phylum Actinobacteria characterized by high cytosine and guanine contents in DNA. Currently, this genus included about 145 different species [2]. More than half of the species were isolated from human and animal clinical samples which indicates their potential participation in pathogenesis [1][2][1,2]. In addition, strains of medical and veterinary importance, of the same species, such as Corynebacterium glutamicum and Corynebacterium efficiens, have biotechnological applications [1][2][1,2].
The most important human pathogen is Corynebacterium diphtheriae, which is the etiological agent of diphtheria, a serious, potentially fatal infection of the respiratory tract and occasionally the skin and other mucous membranes such as, e.g., eye, ear, and genital tract. The infection often causes complications in other body organs [3]. Since Friedrich Löffler’s isolation of toxin-secreting C. diphtheriae in 1884 [1], the species has been the best-known and probably most genetically diverse species of the genus [4][5][4,5]. Classical diphtheria is caused by the production of diphtheria toxin (DT) during infections by isolates holding the toxin gene. DT is the main virulence factor responsible for respiratory, neuro- or cardiopathological symptoms, causing pseudo-membranes, paralysis and cardiac failure [1]. In countries with high anti-diphtheria vaccination coverage, the disease is very rare, but in some regions of Africa and Asia diphtheria is still recognised, with thousands of cases reported annually [3]. The disease can emerge in case of the failed implementation of the recommended vaccination programs or lack of booster doses [3].
Corynebacterium ulcerans was described in 1926 by Gilbert and Steward [6]. The species is closely related to C. diphtheriae and also is able to produce DT. Nowadays, in European countries, C. ulcerans is recognized more frequently as an emerging pathogen associated with diphtheria-like symptoms [7][8][7,8]. Growing numbers of human infections caused by C. ulcerans are the result of zoonotic transmission by contact with animal hosts such as goats, cattle, domestic pigs, dogs, cats and even hedgehogs, monkeys, camels, foxes, squirrels, owls, orcas, otters and water rats [9].
Corynebacterium pseudotuberculosis is the etiological agent of ulcerative lymphangitis in equines, mastitis in dairy cattle, oedematous skin disease in buffalos, or abscesses and caseous lymphadenitis (CLA) in small ruminants, such as goat and sheep [10][11][10,11]. C. pseudotuberculosis has caused occasional infection in farm and animal health workers who remain in close contact with infected animals or their raw products, resulting in swellings of the lymph nodes in the neck or groin. C. pseudotuberculosis animal diseases cause severe economic losses [12]. The bacterium was first described in 1888 by Edmond Isidore Etienne Nocard and classified as C. pseudotuberculosis in 1918 by Eberson [13]. Historically, it is the third species known to be able to produce DT. However, toxin-producing C. pseudotuberculosis has been isolated extremely rarely.
Among the pathogenic species of the genus Corynebacterium, the C. diphtheriae, C. ulcerans and C. pseudotuberculosis, which can produce DT, were clustered together in the group of toxigenic corynebacteria named “C. diphtheriae complex” [14].
However, the infections caused by potentially toxigenic corynebacteria have recently changed. Toxigenic C. ulcerans has been isolated from clinical samples more often than in preceding years. Serious invasive infections caused by nontoxigenic C. diphtheriae have been noticed in many countries with high anti-diphtheria vaccination coverage [15]. What is more, new species capable of producing DT were described in 2020, called C. rouxii and C. silvaticum [16]. Based on the genomic sequencing and biochemical and chemotaxonomic analyses, the name of C. belfantii was proposed for strains previously considered C. diphtheriae biotype belfanti [17].
2. The Structure of Diphtheria Toxin
Diphtheria toxin (DT) is encoded by a 1683-base-pair structural gene-tox (NCBI Reference Sequence: NC_002935.2), encoded not on the bacterial chromosome, but by a lysogenic phage called corynebacteriophage beta or corynephage β [18][19][18,19]. The integration of the corynephage β can occur at two specific sites: attB1 and attB2 [20]. Although tox is of bacteriophage origin, the regulation of its expression is reliant on bacteria [21]. The diphtheria toxin repressor gene (dtxR) is present on the bacterial chromosome. Its protein product (DtxR) is able to bind to the tox operator, blocking the transcription [22][23][22,23]. Interestingly, DtxR is activated by heavy metals, especially iron ions [23][24][25][23,24,25]. In the absence of iron ions apo-DtxR exists as an inactive monomer that is in weak equilibrium with a dimeric form. Once activated by the metal ions, DtxR forms stable dimers and two pairs of dimers have been shown to bind to almost opposite faces of the tox operator sequence. DtxR is composed of two major structural domains linked by a flexible tether containing a proline-rich region. The N-terminal domain contains the ancillary and primary metal ion-binding sites, a canonical helix-turn-helix DNA-recognition motif, and an extensive hydrophobic surface necessary for the formation of stable dimers. After DtxR dimers binding to tox promoter, the transcription of tox is repressed. When iron is limiting, the uncomplexed form of DtxR is unable to bind DNA, leading to the induction of diphtheria toxin [20][21][20,21]. The mature extracellular DT is a polypeptide consisting of 535 amino acid residues with a molecular mass of approximately 58 kDa [26][27][28][26,27,28]. The gene sequence analysis indicates that DT is preceded by 25 residues of leader peptide, which is most likely involved in toxin secretion [19]. DT is produced as a proenzyme that requires specific activation for its toxic function, either prior to or immediately after binding to a sensitive cell [29]. It is based on a proteolytic cleavage which cleaves the peptide bond located at the arginine (Arg) residue: Arg190, Arg192, or Arg193, resulting in the formation of two polypeptides: (1) a 193-residue amino-terminal fragment A (DT-A) which corresponds with the catalytic domain of DT, and (2) a 342-residue carboxyl-terminal fragment B (DT-B), corresponding with the translocation and receptor-binding domains of DT [26][30][26,30]. Both fragments remain covalently bound by the disulphide bonds between Cys186 and Cys201, and a reduction of this binding results in free forms of DT-A and DT-B, capable of infecting target cells [27]. DT has been described as the first example of group A–B toxins in which the catalytic and receptor-binding functions are separated into two different polypeptides [31]. Currently, the A–B motif is well-known and almost universal among intracellular toxins [32][33][32,33]. The model of DT structure has evolved over the years, along with the methods available for its determination [29][34][35][29,34,35]. It is assumed that a single DT molecule has three distinct folding domains – C, T and R (Figure 1), symbols of which are derived from the three main functions of this toxin, respectively: catalysis, translocation and receptor binding, respectively. They are arranged in the shape of the letter Y, with the lower part being the T domain, and the upper elements consisting of the C and R domains. The T domain is formed by the α-helical bundle, the R domain by the flattened β-barrel, and the C domain in turn is a combination of structures α and β. Functionally, the C domain forms fragment A of the mature DT, and the T and R domains – fragment B [29][35][36][29,35,36]. Concurrently, a crystallographic analysis showed that in the discussed Y shape, the cleft in the active site of the C domain is blocked by the R domain from accessing the substrate. This is the reason why those two fragments must be separated from each other in order to be toxic [29]. However, no trypsin-sensitive loop was found between the C and T domains that might be responsible for proenzyme proteolysis. Probably this place is created dynamically, which makes detecting it impossible [31]. Furthermore, there was an emphasized high similarity of the T domain to the hydrophobic N-terminal domain of the B chain of the non-toxic protein CRM45, that the ability to form pores in membranes under acidic conditions was attributed to [37][38][37,38].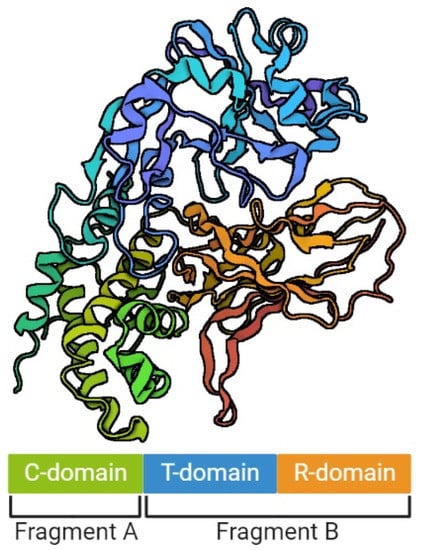
Figure 1.
The structure of diphtheria toxin. The figure was created with
, accessed on 1 September 2022.