Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Brandon Liam Ramchatesingh and Version 2 by Conner Chen.
In the early 20th century, retinol (commonly known as vitamin A) was isolated and characterized as an essential nutrient for human health. Retinoids are natural and synthetic vitamin A derivatives that are effective for the prevention and the treatment of non-melanoma skin cancers. The effects of retinoid signaling on skin physiology have been studied extensively.
- retinoids
- chemoprevention
- chemotherapy
- cutaneous squamous cell carcinoma
1. The Biological Effects of Retinoids in the Skin
The effects of retinoid signaling on skin physiology have been studied extensively [1][90]. Retinoids influence diverse aspects of the biology of epidermal keratinocytes, dermal fibroblasts, skin-resident immune cells, and vascular endothelial cells [2][59]. Examples of processes modulated by retinoids include cell turnover, differentiation, barrier functions, immunity, vascular remodeling and wound healing [3][4][5][6][7][57,91,92,93,94]. The activities of retinoids in healthy skin are harnessed to repress cancer formation.
2. Retinoids Control Epidermal Maturation and Turnover
Retinoids stimulate the process of epidermal turnover [3][57]. Epidermal turnover is the homeostatic tissue renewal process whereby differentiated keratinocytes (corneocytes) undergo desquamation, and are simultaneously replaced by keratinocytes that differentiate and migrate from basal to superficial layers [8][9][95,96]. This process is a delicate balance between basal cell proliferation, differentiation, cell death by cornification and desquamation [9][96]. This effect of retinoids is important for the treatment of diseases of keratinization such as psoriasis, lichen planus, various ichthyosis and to ameliorate fine wrinkling of the skin.
Physiological retinoid signaling inhibits proliferation of epidermal keratinocytes. Ablation of retinoic acid acid receptors (RARs) or transport proteins in epidermal keratinocytes, either in vitro or in vivo, results in basal keratinocyte hyperproliferation, while overexpression promotes proliferation arrest [10][11][12][13][97,98,99,100]. In contrast, high-dose, pharmacological retinoid signaling promotes keratinocyte proliferation. Topical or systemic retinoids, including precursors such as β-carotene or retinaldehyde, induced epidermal hyperplasia in animal models by increasing the thickness of the spinous and granular layers of the epidermis [14][15][16][101,102,103] These effects have been observed in vitro and in vivo, in human and animal subjects, treated with several types of retinoids [15][16][17][18][19][20][102,103,104,105,106,107]. Upregulation of heparin-binding epidermal-growth factor-like (HB-EGF) signaling and activation of fatty acid binding protein 5 (FABP5)/peroxisome proliferating activating receptor β/γ (PPAR β/γ) signaling have been proposed as mechanisms underlying the growth-promoting effects of high-dose retinoids [21][22][23][66,108,109]. On the other hand, several studies provide compelling evidence that high-dose retinoids can induce growth arrest [19][24][25][26][27][28][29][106,110,111,112,113,114,115]. Several growth suppressive mechanisms have been demonstrated, including the activation of tumor suppressor genes like tazarotene-inducible gene 3 (TIG-3), induction of DNA damage and S phase cell cycle arrest, and repression of proliferation-promoting signal transducer and activator of transcription (STAT)STAT signaling [27][28][29][113,114,115]. The discrepancy between these experimental observations has been attributed to context dependency. Retinoid signaling acts in concert with many different signals to either support proliferation or induce arrest [30][116]. As such, retinoids have been proposed to amplify the effect of other signals. Differences between experimental models (e.g., cell lines) and agonist-specific effects (e.g., pathways induced by specific compounds) can also determine the response to retinoids.
Retinoids modulate epidermal differentiation. Animal models and human cases of retinol deficiency provided the first evidence of this [31][117]. Squamous metaplasia, keratinization defects and other skin abnormalities were reported in cases of retinoid deficiency [31][32][3,117]. In embryogenesis and throughout life, a physiological level of retinoid signaling regulates epidermal differentiation [11][98]. Disturbances in the retinoid signaling pathway impair epidermal differentiation [11][33][34][98,118,119]. Both in vitro and in vivo, pharmacological doses of retinoid treatment suppress various stages of epidermal differentiation. Early stage differentiation is impaired, as indicated by the repression of basal cytokeratins 5 and 14, as well as suprabasal keratins 1 and 10 [5][35][92,120]. The final stages of epidermal differentiation, during which time the cornified envelope forms, is also suppressed [36][37][38][121,122,123]. Expression of mucosal keratins (keratin 13) and luminal keratins (keratin 18) are induced by retinoid treatment [35][39][120,124]. It has been proposed that high doses of retinoids may support a mucosal differentiation program while suppressing a squamous differentiation program [39][124]. Mechanistically, this repression may involve regulation of p63, a central transcription factor implicated in terminal differentiation of squamous epithelia [40][125]. Δp63α is the predominant isoform of p63, that is expressed in basal layer keratinocyte stem cells [41][126]. As epidermal differentiation progresses, Δp63α expression in keratinocytes decreases [42][127]. Retinoic acid treatment can inhibit this downregulation, preventing the differentiation-associated decrease in this transcription factor [43][128]. The impact of retinoids on differentiation likely acts in concert with other signals [30][116]. For example, retinoids stimulate synthesis of terminal differentiation genes when applied to differentiating keratinocytes cultured in the presence of high calcium in vitro [44][129]. These properties make retinoids invaluable in the treatment of a variety of papulosquamous conditions affecting the skin/mucosa (lichen planus, psoriasis, Darier disease, etc.) and acquired/inherited disorders of keratinization (ichthyoses, palmoplantar keratodermas, etc.)
Retinoid-induced keratinocyte apoptosis is documented and involves both canonical and non-canonical signaling pathways [45][46][130,131]. Keratinocytes treated with all trans retinoic acid (ATRA) in vitro undergo apoptosis, increasing expression of p53 as well as the expression of caspase 3, 7, 8 and 9 mRNAs and proteins [45][46][130,131]. Other apoptosis-related factors upregulated by retinoid signaling include Fas and BH3 interacting domain death agonist (BID) [5][92]. Beyond the transcriptional effects, Louafi et al. reported that the induction of keratinocyte apoptosis involved nongenomic inhibition of insulin-like growth factor II (IGF2) signaling [47][132]. Selective RAR- and retinoid x receptor (RXR)-specific retinoids are capable of inducing keratinocyte apoptosis [46][131]. Tazarotene promotes apoptosis of immortalized keratinocytes via transcriptional upregulation of p73, a tumor-suppressive p53 homologue [48][133]. Although cell death is intimately tied to terminal differentiation in the skin and the process of cornification, the induction of apoptosis by retinoids is likely independent of the terminal differentiation program. In one study, retinoids repressed expression of keratinocyte differentiation markers while concurrently inducing apoptosis under differentiating culture conditions [46][49][131,134]. Caspase-14, believed to be a central effector of cornified cell death and terminal differentiation, is also repressed by retinoid treatment in murine skin even as cells undergo apoptosis [50][51][135,136]. This may suggest that the apoptotic program initiated by retinoid signaling is independent of the epidermal differentiation program.
The complex, and at times opposing, effects of retinoids on epidermal keratinocytes may be summarized as follows. A physiological level of retinoid signaling is necessary for suppressing proliferation and promoting normal differentiation/keratinization. This physiological level of retinoid signaling is tumor suppressive. Pharmacological doses of retinoid signaling act in concert with other signals within the skin. High doses of retinoids are shown to (1) promote proliferation and epidermal thickening (2) inhibit squamous cell differentiation programs and cornification and (3) promote apoptosis. As highlighted above, these effects make retinoids useful for the treatment of keratinization disorders, keratinocyte carcinoma (KC) and other cancers and their precursor lesions. The reported contradictory effects of retinoids on proliferation brings into question the use of these compounds as therapeutic agents. Nonetheless, it is evident that retinoids do exert an anti-tumorigenic effect within the skin. This may involve a predominance of anti-tumorigenic effects (e.g., apoptosis and cell cycle arrest) over the pro-tumorigenic effect (e.g., proliferation). One mechanism that has been proposed is the predominance of RAR-RXR pathway (anti-tumorigenic) over the PPARβ/γ-RXR pathway (pro-tumorigenic) [21][66]. However, this explanation has been controversial. Another explanation is that the microenvironment within the skin, which cannot be replicated in its entirety in an experimental model, provides a molecular context that supports retinoids’ anti-tumorigenic effects. Importantly, dermatology patients receive retinoid treatments for years and decades (e.g., acitretin for psoriasis treatment) without a documented increased risk of malignancies.
3. Retinoids Influence the Immune Landscape of the Skin
Innate immune effectors within the skin, including dendritic cells and Langerhans cells, are subject to regulation by retinoids [52][53][137,138]. Topical ATRA treatment prevents decrease in Langerhans and dendritic cell density in murine skin exposed to ultraviolet (UV) radiation or chemical carcinogens [53][138]. Skin resident and skin-homing T lymphocytes are also subject to regulation by retinoids. Retinoids promote apoptosis of T lymphocytes and can regulate how circulating lymphocytes home to the skin by regulating the expression of cell-surface adhesion molecules in the epidermis and the infiltrating T cells [54][55][139,140]. The detailed immune modulating effects of retinoids are useful in the treatment of inflammatory skin diseases such as acne and rosacea.
In addition to modifying cellular immunity within the skin, retinoids can support cell-intrinsic immune defenses by limiting replication of some viruses. Human Papilloma Virus (HPV) infection can induce the formation of cutaneous squamous cell carcinomas (cSCCs), driven by the E6 and E7 viral oncoproteins [56][141]. In keratinocytes infected with HPV-16, retinoid treatment repressed transcription of E6 and E7, impairing transformation [57][58][59][60][142,143,144,145]. Additionally, HPV-16 infected keratinocytes were sensitized to retinoid-induced growth arrest and keratinization changes compared to HPV-negative keratinocytes [57][142]. Likewise, retinoids inhibit replication of Kaposi's Sarcoma Herpesvirus (KSHV), the primary causative agent in Kaposi's Sarcoma (KS), in endothelial and epithelial cells in vitro [61][146]. Furthermore, retinoids support healthy barrier defenses, by regulating lipid synthesis and junctional complexes [62][147]. Trifarotene, for example, drives the expression of aquaporin-3 and peptidyl arginase deaminase, which support skin hydration and the integrity of the skin barrier [63][148].
4. Skin Structure and Vascularization Are Regulated by Retinoid Signaling
The complex architecture of the skin is also modified by retinoids. In keratinocytes, retinoids regulate the activity of enzymes, such as collagenases and components of the urokinase plasminogen activator system, which function to inhibit matrix metalloproteases (MMP) [64][65][149,150]. Recently, trifarotene has been shown through gene expression studies to decreased expression of MMP genes in keratinocytes [63][148]. Retinoid signaling also drives turnover of intercellular adhesions, including claudins and corneodesmosomes [62][66][147,151], thereby permitting mobilization of keratinocytes within the epidermis and allowing for eventual desquamation.
Within the dermis and hypodermis, retinoids control vascularization. Retinoids decrease the expression and secretion of vascular endothelial growth factor (VEGF) by keratinocytes [67][152]. By decreasing VEGF produced by the keratinocytes, retinoids restrict dermal angiogenesis [67][68][152,153]. ATRA pre-treatment can also attenuate UV-induced VEGF production by keratinocytes, attributed to the downregulation of the MAPK (mitogen activated protein kinase)/ERK (extracellular signaling regulated kinase) pathway [68][153].
These mechanisms (summarized in Figure 13) allow for retinoids to serve as effective chemoprophylactics and chemotherapeutics. Retinoids are master-regulators of cutaneous physiology, assuming control over processes that are integral to cancer pathogenesis.
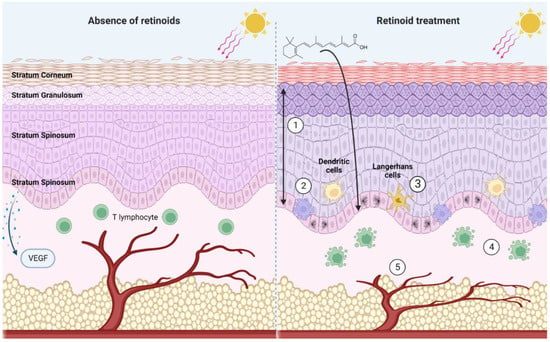
Figure 13. A summary of noteworthy effects of pharmacological retinoid signaling within the skin. In the absence of retinoids, UV radiation depletes the skin of Langerhans cells and dendritic cells. VEGF production by the basal keratinocytes is stimulated by UV radiation, stimulating angiogenesis. In the presence of retinoids (1) Upregulation of basal cell proliferation results in thickening of stratum spinosum and stratum granulosum, and a thinning of the stratum corneum. Cumulative increase in epidermal thickness (2) increased apoptosis of basal cells (3) dendritic cells and Langerhans cells are protected from UV-induced decrease in cell number (4) apoptosis of T cells (5) prevent VEGF release in response to UV irradiation, inhibiting angiogenesis. Created with BioRender.com.