Adipogenic transcription factors including CCAAT/enhancer binding protein alpha (C/EBPα), C/EBP beta (C/EBPβ) and peroxisome proliferator-associated receptor gamma (PPARγ) are also expressed by osteoclastogenic cells. However, in contrast to MSCs, activation of these adipogenic transcription factors in HSCs promotes the differentiation of osteoclast precursors into mature osteoclasts. Herein, we discuss the molecular mechanisms that link adipogenic transcription factors to the osteoclast differentiation program.
- adipocyte osteoclast cross talk ppar gamma
- cebp alpha
- cebp beta.
1. PPARγ: The Master Adipogenic Transcription Factor
A key finding in the field of osteoclast development was that among all the hematopoietic stem cell and progenitor populations in bone marrow, those that express PPARγ are specifically endowed with the ability to commit into the osteoclast lineage[1]. Using in vivo lineage tracing strategies with PPARγ-tTA TRE-H2BGFP reporter mice, Wei et al. (2011) demonstrated that osteoclasts are derived from PPARγ-expressing cells of the hematopoietic lineage[1]. The GFP+ PPARγ+ but not the GFP- PPARγ- bone marrow cells derived from the PPARγ-tTA TRE-H2BGFP reporter mice differentiated into mature osteoclasts when cultured under osteoclastogenic conditions indicating that the osteoclastogenic precursor population lies within the PPARγ+ bone marrow subpopulation. Further, the impact of PPARγ-expressing hematopoietic subsets on bone homeostasis was confirmed by selective depletion of PPARγ+ cells in mice[1]. This was achieved using a novel transgenic mouse model generated by crossing transgenic mice expressing Cre recombinase under the control of the PPARγ promoter driven tetracycline-controlled transactivator protein (tTA) combined with a tetracycline-responsive promoter element (TRE)-Cre transgene (PPARγ-tTA TRE-Cre) with a second line of transgenic mice that exhibited conditional expression of diphtheria toxin (DTA) in the presence of Cre (DTA-floxed). This resulted in the selective ablation of PPARγ+ progenitors, ablation of osteoclast differentiation and an osteopetrotic (abnormally dense bone) phenotype in the offspring[1]. Furthermore, specific activation of Notch in the PPARγ+ progenitor cell populations by breeding the PPARγ-tTA TRE-Cre mice with a constitutively active Notch Intracellular Domain (NICD)-floxed mice suppressed osteoclast differentiation. This is consistent with a role for Notch activation in the suppression of PPARγ signaling in osteoclastogenic cells[1] that is analogous to the suppression of adipogenesis in MSCs by Notch[2][3][4][5][6]. Taken together, these studies indicate that PPARγ activation can induce adverse effects on skeletal homeostasis through the combined effects of reduced bone formation due to suppression of MSC osteoblastogenesis on one hand, and exacerbated bone resorption due to increased HSC osteoclastogenesis on the other.
A similar association exists between the molecular mechanisms that regulate the expression and function of PPARγ. For instance, several studies suggest that the class II histone deacetylase 9 (HDAC9) acts in a negative feedback loop with adipogenic transcription factors such as PPARγ to regulate MSC adipogenesis by repressing PPARγ expression in the undifferentiated state of MSCs, whereas under adipogenic conditions, the stimulation of PPARγ expression transrepresses[2][7][8] HDAC9 gene expression to permit adipogenic gene expression and differentiation[2][7][8]. In parallel to a role in MSC adipogenesis, it was recently reported that HDAC9-knockout (-/-) mice exhibit low bone mass due to exacerbated osteoclast formation and bone resorption[9]. This phenotype could be rescued by transplanting bone marrow from wild-type (WT) mice to HDAC9 (-/-) mice, while transfer of bone marrow from HDAC9 (-/-) mice to WT mice induced the bone resorption phenotype in the latter[9]. This supports the view that the interplay between PPARγ and HDAC9 is relevant to both bone marrow osteoblastogenic and osteoclastogenic cell populations. Further, it is well-established that PPARγ and Wnt/β-catenin form a negative feedback loop in MSCs such that PPARγ drives adipogenesis by transrepressing HDAC9, cyclin D1 and Notch expression and signaling that ultimately limits osteoblastogenic Wnt/β-catenin signaling[2][10]. Parallel to MSCs, it has been reported that PPARγ also downregulates Wnt/β-catenin signaling in HSCs, which in turn induces peroxisome proliferator-activated receptor-gamma coactivator-1 beta (PGC1β) expression during osteoclastogenesis (Figure 1). Mechanistically, PGC1β exhibits dual roles in HSCs by transactivating c-fos expression which induces osteoclast differentiation and by stimulating mitochondrial biogenesis which supports osteoclast function during bone resorption (Figure 1). As such, PGC1β serves as a common mediator for both PPARγ stimulation of osteoclastogenesis and the resorptive functions of mature osteoclasts[1]. In addition, PPARγ also directly induces the expression of estrogen receptor-related receptor alpha (ERRα) which coordinates with PGC1β to induce mitochondrial biogenesis (Figure 1) that additionally contributes to the PPARγ-mediated activation of mature osteoclasts[1]. Consistent with this, targeted deletion of PGC1β in osteoclastogenic cells by breeding PGC1β-floxed mice with Tie2cre-transgenic mice resulted in the abrogation of PPARγ-induced bone resorption[1][11]. Thus, it is now clear that PPARγ is also an integral component of the core transcriptional machinery of HSCs (Figure 1) and activation of this transcription factor under adipogenic conditions in bone marrow can induce osteoclast differentiation and increase bone resorption in disease conditions.
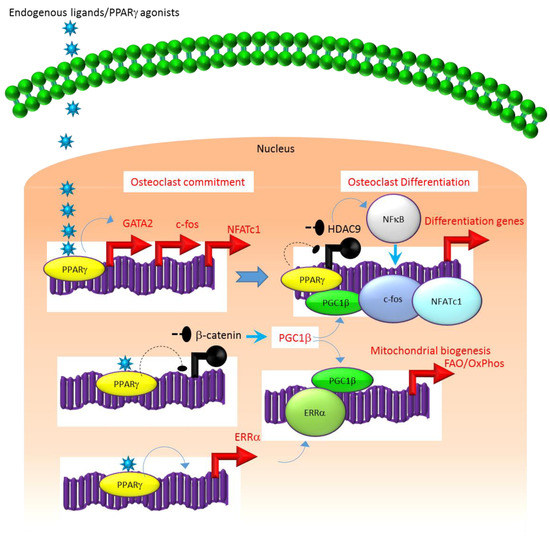
Figure 1. Regulation of osteoclastogenesis by PPARg. Adipogenic stimulus prevailing in bone marrow can induce osteoclastogenesis by activating PPARg that in turn activates the transcription of target genes such as GATA2, c-fos, NFATC1 and ERR1a that induce osteoclast differentiation in HSCs. Additionally, suppression of Wnt/catenin signaling by PPARg can induce PGC1b which can function as a transcriptional co-activator for ERR1a to promote the induction of mitochondrial fatty acid b-oxidation and oxidative phosphorylation genes that are critical for inducing mitochondrial biogenesis to support osteoclast function. PGC1b also can serve as a transcriptional co-activator for PPARg to induce target genes that stimulate osteoclast differentiation. Blue colored arrow-ended lines: stimulation/activation; Black circle-ended dash lines: inhibition. Red colored pointed arrows: transcriptional activation; Black colored closed circles: transcriptional repression.
2. C/EBPα
Similar to PPARγ, considerable experimental evidence also supports a role for the adipogenic transcription factor C/EBPα as a hematopoietic transcription factor[12][13] that promotes the osteoclastogenic differentiation program[14][15][16][17].
Mechanistically, C/EBPα serves as an osteoclastogenic factor through its ability to bind DNA and activate the transcription of several osteoclast-specific transcription factors including NFATc1 and c-fos (Figure 2) as well as genes required for osteoclast resorptive function including cathepsin K (Ctsk) and osteoclast-specific vacuolar proton pump (Atp6i, also known as Tcirg1; Figure 2) that promotes osteoclast differentiation and function[15]. Promoter activity mapping and chromatin immunoprecipitation (ChIP) assays have identified cis-regulatory elements (CREs) located in the promoter regions and have identified these as direct regulatory targets of C/EBPα[15] (Figure 2). This has been linked to paracrine osteoclastogenic signaling by the observation that the 535IVVY538 (IVVY) motif of RANK stimulates C/EBPα expression, which in turn activates the expression of osteoclastogenic genes (Figure 2) and promotes osteoclast differentiation of HSCs[17]. Consistent with this, mutation of the IVVY motif blocked RANKL stimulation of C/EBPα expression and osteoclast differentiation of HSCs[17]. It has been reported that C/EBPα has a substantial and wide-ranging role throughout multiple phases of osteoclast development and function. For example, overexpression of C/EBPα alone was reported to be sufficient to promote osteoclast lineage commitment of HSCs even in the absence of RANKL stimulation[15][17]. Furthermore, ectopic expression of C/EBPα in differentiated macrophages reprogrammed the cells to fuse and convert into osteoclast-like cells with a marked induction of several osteoclast marker genes including TRAP, Ctsk, NFATc1, c-fos, MMP9, TRAF6 and RANK coincident with a downregulation of the macrophage marker F4/80[17]. Consistent with these findings, C/EBPα+/- and C/EBPα-/- mice were reported to exhibit a gene dosage-dependent loss of osteoclast numbers and an osteopetrosis phenotype[15]. In addition to a role in osteoclast lineage commitment and maturation, C/EBPα is also critical for osteoclast function as it is essential to stimulate the extracellular acidification process and to maintain osteoclast survival during resorption[14]. Together, these studies provide evidence that C/EBPα is an osteoclastogenic transcription factor that can effectively induce osteoclast differentiation and function (Figure 2).
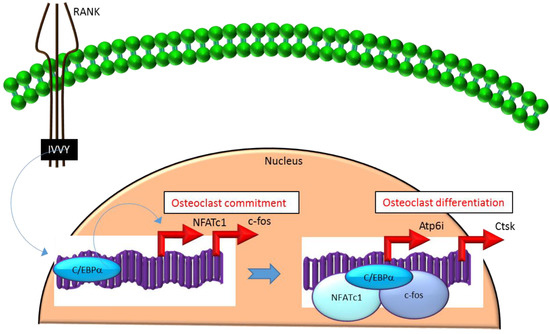
Figure 2. C/EBPa promotes lineage commitment, differentiation and function of osteoclasts. An increase in C/EBPa expression in HSCs is mediated through activation of IVVY motif of the cytoplasmic domain of RANK. C/EBPa can activate transcription of the master osteoclastogenic transcription factor NFATc1 and c-fos to promote osteoclast lineage commitment independent of the presence or absence of RANKL. In addition, C/EBPa can also function as a transcriptional co-activator for NFATc1 and c-fos to induce osteoclastogenic target genes and promote osteoclast differentiation. Blue colored arrow-ended lines: stimulation/activation. Red colored pointed arrows: transcriptional activation.
3. C/EBPβ
C/EBPβ is encoded by an intronless gene that directs the production of four protein isoforms—a 38 kDa full-length C/EBPβ, 34 kDa liver-enriched activating protein (LAP), 21 kDa liver-enriched inhibitory protein (LIP), and a smaller inactive 14-kDa isoform. All of these isoforms are generated by alternative translation initiation from consecutive in-frame start codons directed under the control of the mammalian target of rapamycin kinase (mTOR) pathway[18][19][20][21]. The LIP isoform lacks a major portion of the transactivation domain but retains the DNA-binding domain and dimerization domain that confers its function as a dominant negative regulator of the transactivator isoforms LAP and full-length C/EBPβ[18][19]. It is well-established that regulation of C/EBPβ mRNA expression and the relative levels and turnover of LIP and LAP isoforms play a major role in adipocyte differentiation[22]. For example, constitutive overexpression of C/EBPβ-LIP suppresses adipogenesis, while that of C/EBPβ-LAP promotes adipogenesis by regulating the expression of PPARγ in MSCs[23][24]. The balance between the expression of long transactivator isoforms (full-length or LAP) and the truncated repressor LIP is determined by the mTOR pathway that acts as a key sensor to integrate the differentiation pathways with the nutrient stimuli[18][25][26]. Although some reports suggest that mTOR activates LAP to induce adipogenesis, other evidence indicates that the DEP domain containing the mTOR-interacting protein (DEPTOR) strongly induces PPARγ and adipogenesis by inhibiting mTOR[27]. This suggests that the mechanisms by which C/EBPβ impacts adipocyte differentiation is highly complex and the nature of mTOR signaling determines the relative function of full-length/LAP versus LIP isoforms in inducing or repressing the differentiation program. Similar to the adipogenic cells, the balance between the long transactivator isoforms (full-length/LAP) and the truncated repressor LIP is also regulated by the mTOR pathway in the osteoclastogenic cells[18][19]. Moreover, the osteoclast differentiation program is influenced by the LAP to LIP ratio that determines the activation/repression of C/EBPβ target genes in HSCs[18][19]. In contrast to adipogenesis, HSC osteoclastogenesis is generally suppressed by LAP and stimulated by LIP.
Mechanistically, these actions in HSCs are mediated through the differential regulation of the downstream transcription factor Maf basic leucine zipper (bZIP) transcription factor B (MafB)[18][19][28]. MafB is a negative regulator of osteoclast differentiation and is a direct target gene of C/EBPβ-LAP[18][19][28]. MafB represses the expression of several osteoclast transcription factors including NFATc1, c-fos and microphthalmia-associated transcription factor (Mitf)[18][28] and thereby, inhibits osteoclast differentiation (Figure 3). Consistent with this, it has been reported that repression of MafB resulting from either targeted deletion of LAP or forced expression of LIP was sufficient to induce osteoclast differentiation in HSCs[19]. Taken together, these studies indicate that the ratio of the transactivator to repressor isoforms of C/EBPβ is a key determinant of HSC fate by linking the mTOR pathway to the osteoclast differentiation program through regulation of MafB expression (Figure 3).
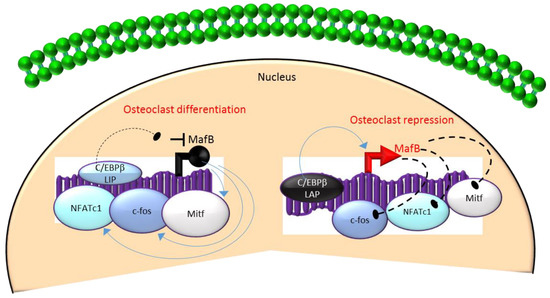
Figure 3. A negative feedback loop between C/EBPb and MafB regulates osteoclastogenesis. C/EBPb is produced as two major isoforms: C/EBPb-LAP (stimulatory) and C/EBPb-LIP (inhibitory) which activate or repress the basic region/leucine zipper transcription factor MafB, respectively. The repression of MafB by LIP induces osteoclast differentiation due to the loss of MafB-mediated repression of NAFTc1, c-fos and Mitf. In contrast, stimulation of MafB expression by LAP blocks osteoclast differentiation due to MafB-mediated repression of NAFTc1, c-fos and Mitf. Blue colored arrow-ended lines: stimulation/activation; Black circle-ended dash lines: inhibition. Red colored pointed arrows: transcriptional activation; Black colored closed circle: transcriptional repression.
This entry if from 10.3390/ijms21072277