Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Jiawei Chen.
There is a wide variety of kinds of lipids, and complex structures which determine the diversity and complexity of their functions. With the basic characteristic of water insolubility, lipid molecules are independent of the genetic information composed by genes to proteins, which determine the particularity of lipids in the human body, with water as the basic environment and genes to proteins as the genetic system. After the well-studied PI3K-AKT pathway, insulin affects fat synthesis by controlling the activity and production of various transcription factors.
- lipid metabolism
- regulation network
- insulin
1. Introduction
Lipids are the general name of fats and lipoids. Fats are triglycerides (TG); lipoids include cholesterol and its esters, phospholipids, glycolipids. Triglycerides play an important role in energy storage and supply. Exogenous triglycerides are mainly obtained from food, while endogenous triglycerides are mainly synthesized by the liver, adipose tissue, and small intestine. In the liver, they are assembled with apolipoproteins, phospholipids and cholesterols to form very low-density lipoprotein (VLDL) and then is transported out of the liver. Disorder of VLDL synthesis will lead to the accumulation of triglycerides in liver cells—namely fatty liver. Because the basic substrates for fatty acid synthesis, such as acetyl coenzyme A, ATP and NADPH are mainly derived from glucose catabolites, triglyceride metabolism is associated with glucose metabolism. In addition to the metabolic changes in the synthesis decomposition balance caused by changes in substrates supply, hormones also affect fat metabolism. Hormones like adrenaline, norepinephrine, and glucagon triggered by fasting, starvation and sympathetic excitement stimulate fat mobilization. Cyclization of adenylate further activates cAMP-dependent protein kinase and increases adipose triglyceride lipase (ATGL) activity. However, the key enzyme for fatty acid synthesis, Acetyl-CoA carboxylase, is inhibited. In turn, insulin plays an anti-lipolysis role, which activates acetyl coenzyme A carboxylase to promote fatty acid synthesis, and insulin increase fat storage in adipose tissue. As an important component of TG, cholesterol esters and phospholipids, fatty acid can be used to synthesize strong bioactive substances such as prostaglandin, leukotrienes, and thromboxane A2, who play a role in regulating local immunity. Phospholipids and their derived components are the basic components of biological membranes. In the inner layer of cell membranes, phosphatidylinositol-4,5-diphosphate plays a role in transmitting cell signals. Cholesterol is also an important component of bio-membrane. In some endocrine glands, cholesterol is used to synthesize steroid hormones. However, high concentration of cholesterol in the blood can induce atherosclerosis. For cholesterol synthesis, HMG-CoA reductase is a crucial enzyme. Insulin and thyroid hormones induce the synthesis of HMG-CoA reductase to promote the process, while thyroid hormones promote the conversion of cholesterol into bile acid. Glucagon chemically modifies and regulates the phosphorylation of HMG-CoA reductase and inactivates it, while cortisol directly inhibits its activity. In the circulation, blood lipids are mainly transported and metabolized in the form of plasma lipoproteins. Plasma lipoproteins are divided into several main types of lipid and protein contents, with different physiological functions. As mentioned above, VLDL is the main form for transporting endogenous TG, while low-density lipoprotein (LDL) mainly transports endogenous cholesterol. Oxidative modified LDL and VLDL are considered as important pathogenic factors of cardiovascular and cerebrovascular diseases. High-density lipoprotein (HDL) is considered to be a protective factor of cardiovascular and cerebrovascular diseases, due to its ability to transport the peripheral cholesterol to the liver for metabolism.
2. Insulin and Transcriptional Regulation
Insulin plays a central role in lipid metabolism—promoting energy storage and inhibiting energy release; this can be shown especially in insulin-resistant individuals who will develop fat accumulation in the liver, a condition induced by increased de novo lipogenesis [1]. Accumulating studies have shown phosphoinositide 3-kinase (PI3K)-Akt pathway to be a signaling cascade; being integral to the metabolic actions of insulin, after insulin binding to its cognate receptor, PI3K is recruited to phosphorylate insulin receptor substrates (IRS) and generates 3′phosphoinositides. Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) generation promotes the recruitment of pyruvate dehydrogenase kinase 1 (PDK1) and Akt (also known as protein kinase B), leading to the subsequent phosphorylation of Akt by PDK1. This phosphorylation can be also induced by mammalian targeting of rapamycin (mTORC) 2, but within a different amino acid site [2]. Then, multiple downstream pathways such as mTORC1, glycogen synthase kinase and the FoxO transcription factors were signaled to control glucose and lipid metabolism [3,4][3][4].
As the function of AKT in human energy and material metabolism is elucidated, more downstream pathways of Akt are being studied extensively. This includes the sterol regulatory element-binding protein 1c (SREBP1c) and the carbohydrate response element-binding-protein (ChREBP), which belong to lipogenic transcription factors activating lipogenic genes such as Fasn, ACC, Scd1, and Elovl6 [5]. During the synthesis of fatty acids, TGs, cholesterol and its esters, SREBPs are a class of transcription factors that exist widely and play an integral role [6]. Insulin activation of Akt enhances both the synthesis and processing of SREBP1c, which is the predominant SREBP subtype in hepatocytes; in SREBP1c precursor processing, insulin enhances the affinity of the SREBP1c precursor complex for vesicles and Golgi apparatus, and increases proteolytic cleavage of the precursor protein induced by SREBP cleavage-activating protein (SCAP) in an PI3K-AKT-dependent way [7]. The requirement to SREBP1c in some pathological states, including insulin resistance-induced hepatic fat accumulation, has been proved in SCAP-specific knockout mice models [8]. SREBP1c also serves as a connection between lipid and glucose metabolism. Under low ATP supply status, the upregulated AMP-activated protein kinase (AMPK) is present in hepatocytes phosphorylates SREBP1c and thus inhibits de novo lipogenesis (DNL). The activation of AMPK could protect high-fat feeding rodents from hepatocytes steatosis or atherosclerosis [9]. Furthermore, SREBP1c has been shown to play an important role in adipocyte differentiation [10]. ChREBP is a glucose-responsive transcriptional factor. Specific inhibition of ChREBP in mice hepatocytes reduces DNL and TGs [11]. However, it is still elusive whether SREPB1c and ChREBP are activated in insulin-resistant individuals, as increased fat synthesis and storage could be observed in these patients [12]. More extensive studies are needed on the interaction between these transcription factors and their role in the human body.
Similar to SREBPs and ChREBP, FoxO proteins are also transcriptional factors controlled by Akt through a delicate phosphorylation action, which mainly inhibit the expression of lipogenic target genes [13]. The transcriptional mechanism of FoxO1-controlling liver lipid metabolism has not been fully determined. For the SREBP1c promoter, there is evidence which shows that FoxO1 binds to it directly and reduces its transcriptional activity [14]. The glucokinase, as a lipogenic gene that functions by increasing the glucose-6-phosphate level and activating the subsequent ChREBP, is also affected by FoxO1. Based on the current evidence, AKT-dependent inhibition of FoxO1 seems to be required for DNL [15]. Experiments have yielded conflicting results still on the role FoxO1 plays in triglyceride secretion. It was reported that overexpression of FoxO1 in transgenic mouse hepatocytes resulted in elevated ApoCIII and subsequent hypertriglyceridemia [16]. Similarly, FoxO1 was reported to be necessary for the expression of microsomal triglyceride transfer protein (MTP), which led to the secretion of TG-rich VLDL [17]. However, Zhang et al. [18] reported decreased serum TG levels due to the failure to repeat Foxo1-reduced VLDL triglyceride content and ApoCIII-induced serum TG rising in vivo or in vitro. Consistent with this finding, hepatic specific deletion of FoxO1 does not alter serum TG levels. Insulin signaling leads to Foxa2 inactivation through Thr156 phosphorylation and nuclear exclusion, which inhibits Foxa2 target gene expression and decreases hepatic lipid metabolism [19]. Characterization of FoxO6 as an integrator of hepatic insulin signaling with TG-rich VLDL (VLDL-TG) production in the liver provides important insights into the mechanism of hypertriglyceridemia. FoxO6 did not undergo insulin-dependent phosphorylation and nuclear translocation like other FoxO subtypes did [20]. FoxO6 promoted lipogenesis and augmented VLDL-TG secretion, which was demonstrated by Kim and his colleagues [21]. This action correlated with the ability of FoxO6 to stimulate hepatic production of MTP, which is a key to enzyme-promoting VLDL assembly and secretion by catalyzing lipid transferring to nascent apolipoprotein B (apoB) [22]. Although controversies remain on this topic, there is no doubt that FoxO, especially FoxO1, plays an important role in insulin-mediated lipid metabolism. Insulin signaling pathway is summarized as Figure 1.
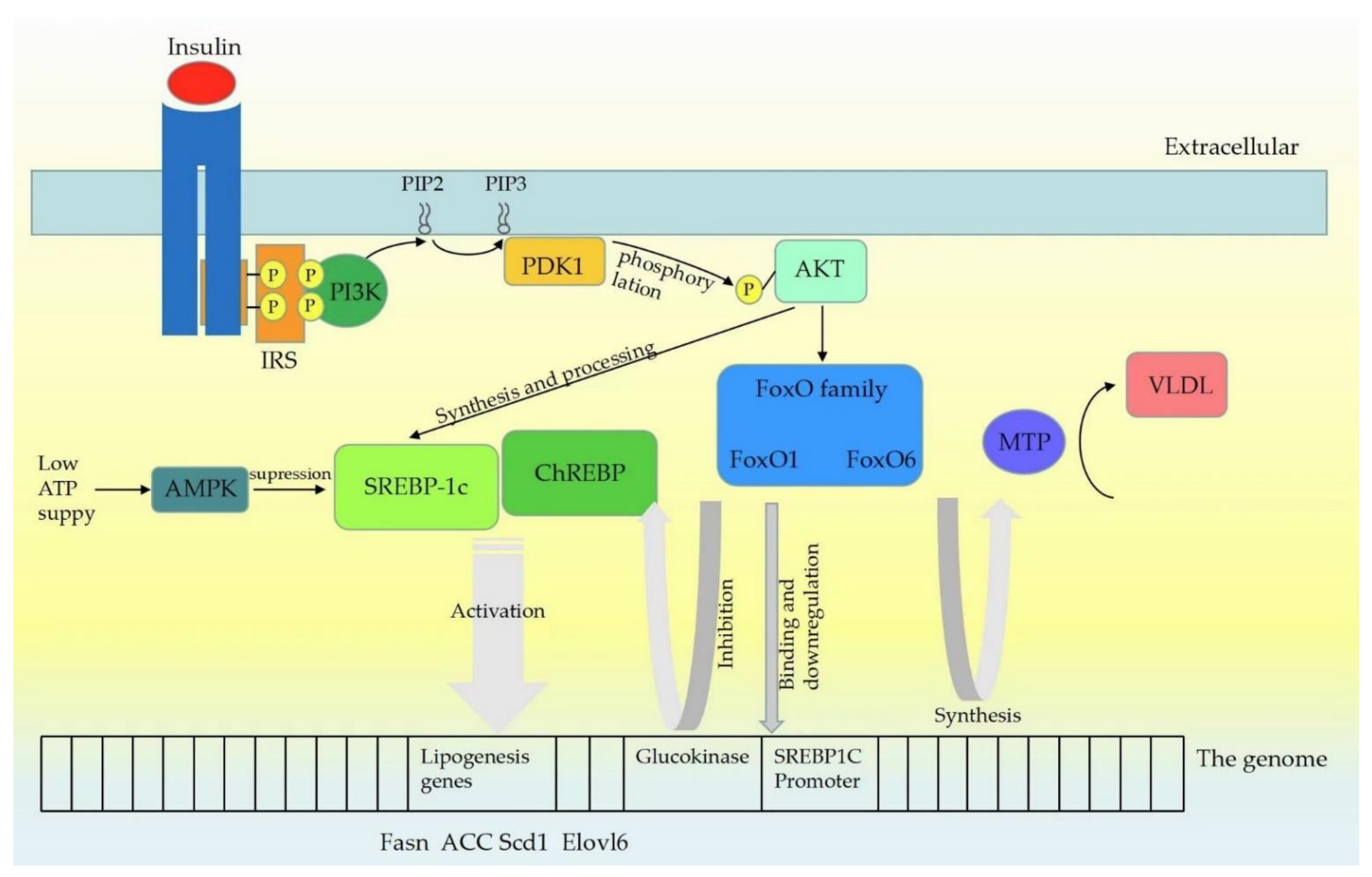
Figure 1. Insulin signaling pathway mechanism. Upon insulin receptors binding to ligands, leading to self-phosphorylation, insulin receptor substrates (IRS) are recruited and phosphorylated. IRS proteins are then recruited and activate PI3K, which phosphorylates PIP2 to generate PIP3. PDK1 is then activated by PIP3, which phosphorylates Akt at Thr308. Insulin activation of Akt enhances both the synthesis and processing of SREBP (SREBP1c mainly) in hepatocytes. SREBP1c and ChREBP activate lipogenic genes such as Fasn, ACC, Scd1 and Elovl6. At low cellular ATP levels, activation of AMPK interacts with and phosphorylates SREBP1c, thus inhibiting proteolytic cleavage and nuclear translocation, and repressing DNL. Similar to SREBP1c, FoxO proteins are also the class of transcription factors downstream of INS signaling pathway, and Akt controls the activity of the FoxO proteins through a phosphorylation mechanism. FoxO1 can bind to the SREBP1c promoter directly and further affect the transcriptional activity of Sp1 and SREBP1c. FoxO1 controls the expression of the glucokinase gene which may promote the lipogenic response by increasing glucose-6-phosphate levels and activating ChREBP. FoxO6 stimulated hepatic production of microsomal triglyceride transfer protein (MTP), which catalyzed the transfer of lipid to nascent apolipoprotein B (apoB), a rate-limiting step in the nascent assembly and secretion of VLDL.
A growing amount of evidence from in vivo studies supported the idea that MTORC1 plays a role in activation of the AKT downstream pathway [23]. Despite the complexity of Akt downstream pathway, the most critical point is the regulation of lipid metabolism and the progression of some abnormal physiological status which requires insulin signaling via Akt.
References
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50.
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96.
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806.
- Lin, H.V.; Accili, D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011, 14, 9–19.
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268.
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131.
- Yellaturu, C.R.; Deng, X.; Cagen, L.M.; Wilcox, H.G.; Mansbach, C.M., 2nd; Siddiqi, S.A.; Park, E.A.; Raghow, R.; Elam, M.B. Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J. Biol. Chem. 2009, 284, 7518–7532.
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246.
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388.
- Klemm, D.J.; Leitner, J.W.; Watson, P.; Nesterova, A.; Reusch, J.E.; Goalstone, M.L.; Draznin, B. Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J. Biol. Chem. 2001, 276, 28430–28435.
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170.
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338.
- Gross, D.N.; van den Heuvel, A.P.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336.
- Deng, X.; Zhang, W.; I, O.S.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143.
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016, 23, 1154–1166.
- Altomonte, J.; Cong, L.; Harbaran, S.; Richter, A.; Xu, J.; Meseck, M.; Dong, H.H. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J. Clin. Investig. 2004, 114, 1493–1503.
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364.
- Zhang, W.; Patil, S.; Chauhan, B.; Guo, S.; Powell, D.R.; Le, J.; Klotsas, A.; Matika, R.; Xiao, X.; Franks, R.; et al. FoxO1 regulates multiple metabolic pathways in the liver: Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 2006, 281, 10105–10117.
- Von Meyenn, F.; Porstmann, T.; Gasser, E.; Selevsek, N.; Schmidt, A.; Aebersold, R.; Stoffel, M. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. 2013, 17, 436–447.
- Kim, D.H.; Perdomo, G.; Zhang, T.; Slusher, S.; Lee, S.; Phillips, B.E.; Fan, Y.; Giannoukakis, N.; Gramignoli, R.; Strom, S.; et al. FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes 2011, 60, 2763–2774.
- Kim, D.H.; Zhang, T.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Piccirillo, A.; Fan, Y.; Uppala, R.; Goetzman, E.; Dong, H.H. FoxO6 integrates insulin signaling with MTP for regulating VLDL production in the liver. Endocrinology 2014, 155, 1255–1267.
- Lee, S.; Dong, H.H. FoxO integration of insulin signaling with glucose and lipid metabolism. J. Endocrinol. 2017, 233, R67–R79.
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446.
More