Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mariusz Mojzych and Version 2 by Vivi Li.
The Mitsunobu reaction plays a vital part in organic chemistry due to its wide synthetic applications. It is considered as a significant reaction for the interconversion of one functional group (alcohol) to another (ester) in the presence of oxidizing agents (azodicarboxylates) and reducing agents (phosphines). It is a renowned stereoselective reaction which inverts the stereochemical configuration of end products. One of the most important applications of the Mitsunobu reaction is its role in the synthesis of natural products.
- natural products
- Mitsunobu reaction
- alkaloids
- lignans
- polyketides
- amino acids
- carbohydrates
1. Introduction
The Mitsunobu reaction is considered as an important reaction because of its great applications in organic synthesis. It was discovered by Oyo Mitsunobu during the mid-twentieth century. It is a dehydrative redox reaction that converts an alcohol to an ester by coupling with an acid/pronucleophile [1]. The pronucleophiles include oxygen pronucleophiles, such as carboxylic acids, nitrogen pronucleophiles, such as imides and sulfonamides, and sulfur pronucleophiles, such as thiols, etc. [2]. This reaction is mediated by phosphines, i.e., triphenylphosphine, and azodicarboxylates, i.e., diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), etc., and a solvent [3]. A general Mitsunobu reaction is represented in Scheme 1.
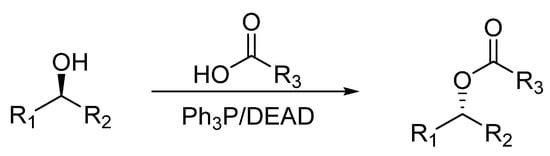
Scheme 1.
General Mitsunobu reaction. Where, R
1
, R
2
, R
3
= Alkyl/Aryl.
In case of sterically hindered alcohols, 4-nitrobenzoic acids or phthalimides are also used for coupling. Besides these, many other reagents, such as fluorinated alcohols and hydroxamates, can also be employed for Mitsunobu coupling [4]. In this way, this reaction allows the formation of C-C, C-N, C-O and C-S bonds formation owing to its diverse selection of reagents. The panoramic feature of this reaction is that it brings about inversion of configuration by coupling of a chiral secondary alcohol and a pronucleophile [5].
The Mitsunobu reaction has a wide range of applications in organic synthesis as it encapsulates the total synthesis of natural products, drugs, analogues, and semisynthetic derivatives of naturally occurring compounds, etc. It constitutes a key step in the synthesis of many of the natural products and other pharmaceuticals. Figure 1 describes a few examples where the Mitsunobu reaction has played its crucial role:
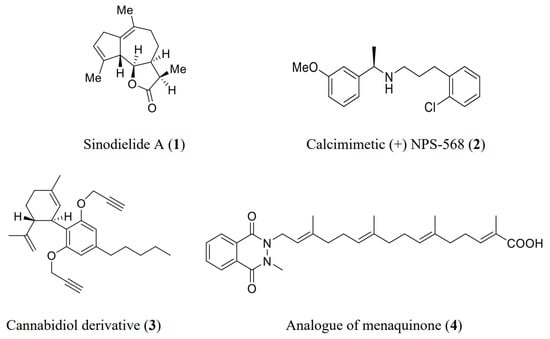
Figure 1.
Structures of some biologically active natural compounds and synthetic derivatives.
The total synthesis of sinodielide A 1, which is used as a flavoring, fragrance, medicine, and poison, employs the Mitsunobu reaction as an important step [6][7][6,7]. Similarly, the synthesis of anti-proliferative menaquinone 4, which is a homologue of vitamin K, and synthesis of cannabidiol derivatives 3, which is used for the treatment of arthritis, immune system disorders, and diabetes, include the Mitsunobu reaction as a key step [8][9][8,9]. It has played a great role in the synthesis of drugs and their derivatives, such as the synthesis of NPS R-568 2, which is a calcimimetic drug used for the treatment of primary and secondary hyperparathyroidism [10].
2. Alkaloids-Based Natural Product Synthesis
2.1. Yuzurimine Alkaloids
The Mitsunobu reaction has been used for the modification and synthesis of scaffolds for the total synthesis of natural products. The yuzurimine alkaloids are extracted from Daphniphyllum macropodum [11] and exhibit many biological activities. In 2019, Hayakawa et al. proposed the synthesis of the heterocyclic portion of yuzurimine-type alkaloids [12]. For this purpose, a protected alcohol 6 synthesized from precursor 5 underwent the Mitsunobu reaction for intramolecular alkylation and gave Ns amide 7 in a 69% yield. The next step involved selective deprotection of hydroxyl group to give 8 in a 75% yield. An intermolecular Mitsunobu reaction was performed to give a bicyclic product 9, which could be converted to the desired product deoxyyuzurimine 10 over a few steps (Scheme 2).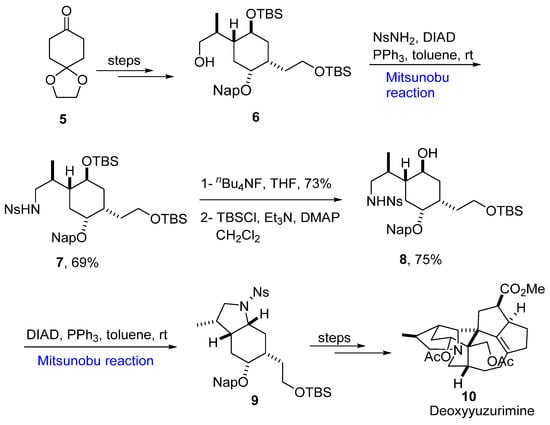
Scheme 2.
Synthesis of deoxyuzurimine alkaloid
10
.
2.2. Phenanthrene Alkaloids
Phenanthrenes are known as chromophores that exhibit many physiochemical properties, such as photoconductance and electroluminescence [13]. Aristolactams are phenanthrene-related compounds that are isolated from Aristolochia argentina. These have been used in many folk medicines in the past [14]. Luong et al. reported the total synthesis of aristolactam 15 in 2019 [15]. To achieve the task an appropriate aldehyde 11 was converted to phenol 12 over a few steps. In the next step, an already synthesized alcohol 13 with an anti:syn ratio of 6:1 was utilized to couple with phenol 12 via a Mitsunobu reaction in the presence of diisopropyl azodicarboxylate (DIAD) and triphenyl phosphine (PPh3) in toluene at 0 °C to 90 °C to obtain 76% of the coupled product 14. The last step included the global deprotection and cyclization of ether by using TFA in anisole that provided stereoselective (+)-aristolactam 15 in a 32% yield (Scheme 3).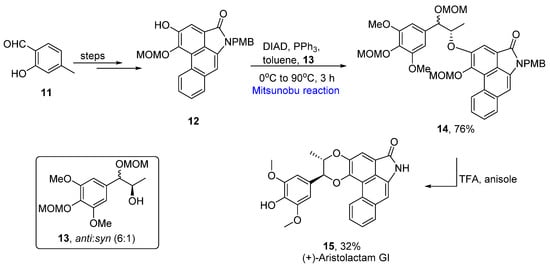
Scheme 3.
Total synthesis of (+)-aristolactam
15
.
2.3. Indole Alkaloids
Nauclefine is an indole alkaloid which was extracted from a West African tree named Nauclea latifola. It exhibits a number of biological activities, such as anti-tuberculotic, anti-inflammatory, and anti-neurodegenerative [16][17][16,17]. In 2021, Chen et al. proposed the total synthesis of nauclefine 24 by using commercially available starting materials [18]. The total synthesis was accomplished by employing the Mitsunobu reaction, Fischer indolization, and allylic oxidation in six steps. In the first step, the TMS-protected alkynol 16 was made to react with the hydroxy isoindolene derivative 17 via a Mitsunobu reaction in the presence of PPh3 and DIAD in THF at 0 °C to room temperature to give alkyne 18 in a 94% yield. In the next step, the alkyne 18 was reacted with hydrazine monohydrate using MeOH/DCM and freshly prepared nicotinic acid in the presence of K2CO3, EtOAc/H2O to yield nicotine amide followed by catalysis in the presence of 2.5 mol% of (Cp*RhCl2)2 as a catalyst and 3 eq. of CsOAc with TFE to furnish regioisomeric products 20 and 21 in the yield of 28 and 69%, respectively. Later on, naphthyridine derivatives were treated via Mitsunobu reaction conditions with PPh3, DIAD, and THF in a reaction mixture to give tricyclic product 23 in a 76% yield. It was further oxidized in the presence of SeO2 and Dess–Martin periodinane followed by Fischer indolization to yield the desired product, nauclefine 24 in a 66% yield (Scheme 4).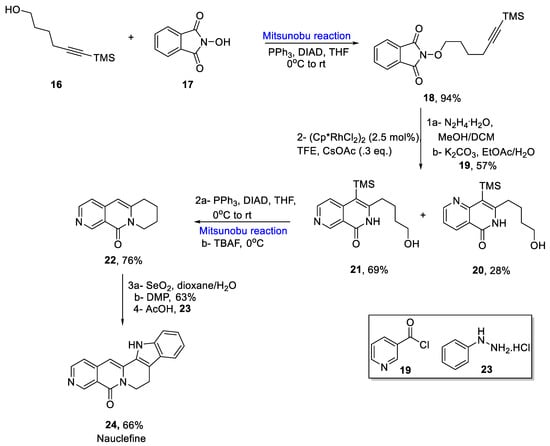
Scheme 4.
Total synthesis of nauclefine
24
.
3. Neolignans-Based Natural Product Synthesis
Plant-based lignans exhibit a number of biological activities, such as antileishmanial, antiparasitic, trypanocidal, and antitumor [19][20][49,50]. Surinamensinols are lignan-based natural products, which exhibit anti-inflammatory and anti-cancerous activities. Surinamensinol A and B demonstrate appreciable anti-cancerous activity against A549, SK-MEL-2, HCT-15, and SK-OV-3 cell lines [21][51]. Various research groups have reported the total synthesis of surinamensinols consisting of lengthy schematic pathways. In 2020, Avula et al. reported an expeditious method for the total synthesis of surinamensinols by using commercially available starting materials consisting of six steps [22][52]. The first step involved the reaction of allyl alcohol 157 and benzyl bromide in NaH and anhydrous THF to give a 98% product, which was further reacted with 9-borabicyclo [3.3.1] nonane solution (9-BBN) in THF, followed by reaction with 4-bromo-2-methoxyphenol 158 in Pd(PPh3) and THF via Suzuki–Miyaura coupling to yield 86% of phenol benzyl ether 159. In the following step, phenyl benzyl ether 159 was made to couple with (S)-ethyl lactate 160 by Mitsunobu reaction in the presence of diisopropyl azodicarboxylate (DIAD), triphenyl phosphine, and anhydrous THF under reflux for 24 h to give the required chiral ester 161 in the yield of 88%. The compound 161 was then reduced by using DIBAL-H, and then subjected to a reaction with 3,4,5-trimethoxyphenyl magnesium bromide to yield a anti and syn diastereomeric mixture of products 162. This mixture of products was then deprotected by using 10%Pd/C and ethyl acetate. The final products 163 and 164 were obtained in overall 14% and 22% yields, respectively (Scheme 5Scheme 21).
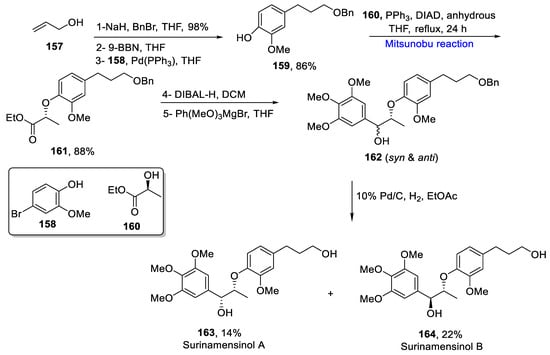
Scheme 521.
Total synthesis of surinamensinols A
163
and B
164
.
Lignans are natural compounds which have a number of medicinal properties, i.e., anticancer, antimicrobial, anti-inflammatory, antiviral, antifungal, and neurotoxic etc. [23][53]. Aglacins are referred to as aryl tetracyclic lactone lignans, which are isolated from Aglaia cordata. Xu et al. proposed the total synthesis of aglacins by employing an asymmetric photoenolization/Diels–Alder (APEDA) reaction as a key step, in 2021 [24][54]. 2-Methyl benzaldehyde 165 and bromo furan derivative 166 were combined to give compound 167 over a few steps, which was reduced in the presence of LiAlH4 to yield 168 in an 88% yield. A Mitsunobu reaction was employed in the presence of triphenyl phosphine and di-tert-butyl-azodicarboxylate to form a cyclic ether ring followed by TIPS deprotection by using tetrabutylammonium fluoride to obtain (−)-aglacin E 169 in a 50% yield. Aglacin E was further converted into (−)-aglacin A 170 in a 49% yield by employing the Mitsunobu reaction and (−)-aglacin B 171 in a 90% yield by reductive dihydroxylation (Scheme 6Scheme 22).
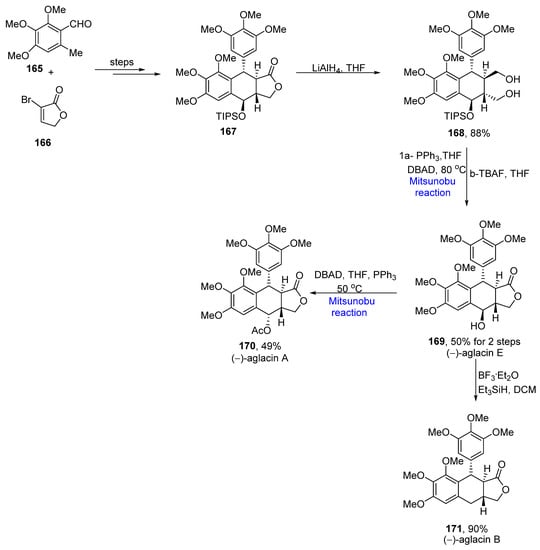
Scheme 622.
Total synthesis of (−)-aglacin A
170
, B
171
and E
169
.
Furthermore, synthesis of (+)-linoxepin 177 commenced from the APEDA reaction of 172 and dienophile 166, which provided 76% of tricyclic product 173 with 93% enantioselectivity. Next, the desired triflate 174 was obtained by oxidation in the presence of DMP and sodium bicarbonate accompanied by triflation in the presence of Tf2O in an 84% yield over two steps. In the following step, a Pd-catalyzed Suzuki–Miyaura coupling reaction between triflate 174 and compound 175 was performed in the presence of Pd(PPh3), 1,4-dioxane and phosphoric acid to obtain the desired coupling product 176 in a 60% yield. The next step involved the removal of TBS and two methyl groups followed by an intramolecular Mitsunobu reaction for seven-membered cyclo-ether ring formation along with the addition of a methyl group in the presence of methyl iodide to obtain the final product (+)-linoxepin 177 in a 40% yield over two steps (Scheme 7Scheme 23).
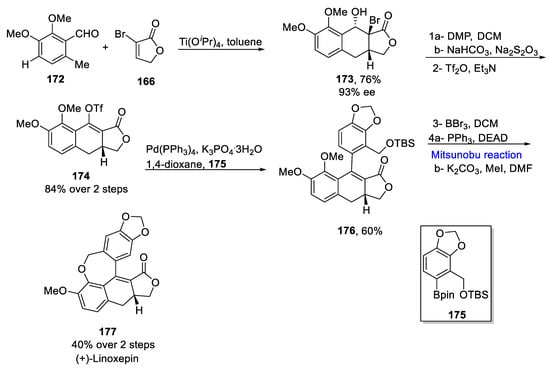
Scheme 723.
Total synthesis of (+)-linoxepin
177
.
Lignans are an important class of natural products that possess a number of biological activities, such as antitumor, anti-inflammatory, anti-bacterial, and immunomodulators. 7S-HMR is converted to (−)-enterolactone by intestinal bacteria and exhibits chemo preventive effects on the development of mammary carcinoma induced by DMBA. Furthermore, it possesses anti-oxidant potential and exhibits chemopreventive effects in an ApcMin mice model of human familial adenomatous polyposis [25][55]. These are present in many fruits, vegetables, and seeds. 7R-Hydroxymatairesinol belongs to lignans which have polyphenolic structures. It is reported to have chemotherapeutic and metabolizing effects. In 2021, Colombo et al. reported the preparation of a 7R-HMR isomer from a 7S-HMR isomer using kinetic reduction, Mitsunobu reaction for epimerization, and ester hydrolysis as key steps [26][56]. In the first step, the protection of phenolic functional groups in the 7S-HMR 178 isomer was completed by using TBSCl and imidazole in DMF to obtain alcohol 179 in a 68% yield. In the next step, a Mitsunobu reaction was performed in the presence of PPh3, DIAD, THF, and p-nitrobenzoic acid at room temperature for 24 h to obtain 180 with an inverted OH group in a 43% yield. The final step involved hydrolysis to ensure complete inversion, followed by deprotection using TBAF and AcOH in THF to obtain 7R-HMR 181 in a 72% yield (Scheme 8Scheme 24).
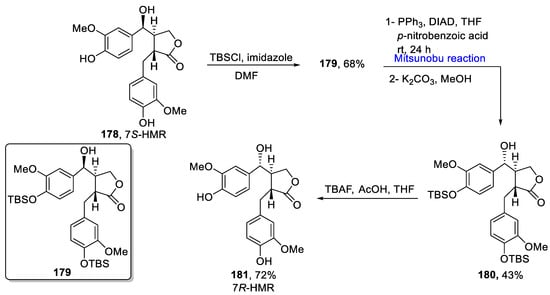
Scheme 824.
Preparation of 7
R
-HMR
181
isomer from 7
S
-HMR
178
.
Neoflavones are naturally occurring compounds and act as important scaffolds for the synthesis of drugs and biologically active compounds. Prenyl groups enhance the bioactivity of respective compounds. They exhibit protease inhibition in COVID-19 [27][57]. Prenylneoflavones are derivatives of neoflavones whose synthesis has been reported by Lozinski et al. in 2020. They employed a Mitsunobu reaction, olefin cross-metathesis reaction, and Claisen rearrangement as key steps to obtain a 5% overall yield in six steps [28][58]. The first step involved the conversion of 3-methoxyacetophenone 182 to neoflavone 183 in a 52% yield via Claisen rearrangement in the presence of diethyl carbonate, and Pechmann condensation in the presence of resorcinol in sulfuric acid. Next, the Mitsunobu reaction with allyl alcohol in the presence of diazo-dicarboxylate (DIAD) and triphenyl phosphine at 0 °C gave 7-allyloxy-neoflavone 184 in a 78% yield. Further, Claisen rearrangement with Eu(fod)3 along with acylation gave an acetylated product 185 in a 79% yield. The 7-acetoxyneoflavone was further treated with Grubbs second generation catalyst to yield prenylneoflavone 186 in a 65% yield (Scheme 9Scheme 25).
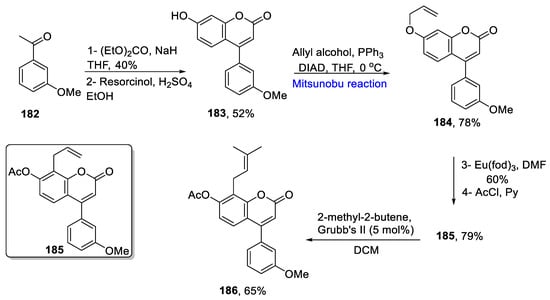
Scheme 925.
Total synthesis of prenylneoflavone
186
.
Lignans are polyphenolic compounds that possess many biological and medicinal properties [29][59]. Ligraminol D and E are lignans which are isolated from Acorus. gramineus and exhibit anti-proliferative activity against tumor cells and inhibit the production of nitric oxide in BV-2 cells [30][60]. Mane et al. proposed the total synthesis of Ligraminol D and E in 2019 [31][61]. The synthesis of Ligraminol E was commenced from known aldehyde 187, which was converted to a chiral diol 188 over a few steps. Next, diol 188 was made to react with benzoyl chloride for the selective protection of the hydroxyl group and production of secondary alcohol 189 in a 90% yield. It was further treated with acetate 190 via Mitsunobu reaction in the presence of DEAD and PPh3 in anhydrous THF under reflux for 6 h to give 88% of ether 191, which was hydrogenated in the presence of Pd/C followed by a global reduction to yield Ligraminol E 192 in a 93% yield (Scheme 10Scheme 26).
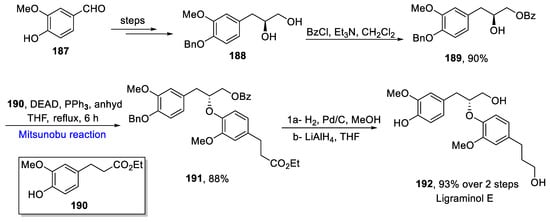
Scheme 1026.
Total synthesis of Ligraminol E
192
.
For the synthesis of Ligraminol D, alcohol 194 obtained from aldehyde 193 over a few steps was made to undergo a Mitsunobu reaction with ester 190 in the presence of PPh3 and DEAD in anhydrous THF under reflux for 6 h to give 195 in an 87% yield. It was converted to Ligraminol D 196 by debenzylation in a 93% yield (Scheme 11Scheme 27).
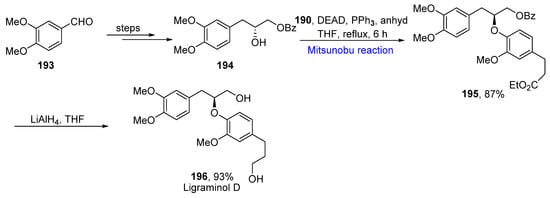
Scheme 1127.
Total synthesis of Ligraminol D
196
.
4. Synthesis of Polyketides-Based Natural Products
Resorcyclic acid lactones are a major class of naturally occurring compounds [32][64]. Paecilomycin A-F are polyketides that belong to resorcyclic acid lactones and are isolated from mycelial culture of Paecilomycessp. They exhibit a number of medicinal and biological properties, such as anti-malarial, antibacterial, anti-enzymatic, anti-parasitic, nematicidal, and anti-fungal. Paecilomycins are found to express anti-plasmodial activity in opposite to Plasmodium falciparum. Among all paecilomycins, paecilomycin E exhibited the best activity with an IC50 value of 20.0 nM [33][65]. In 2019, Reddy et al. proposed the total synthesis of paecilomycin A-F by employing the key steps of Alder–Rickert, Mitsunobu esterification, and ring closing metathesis [34][66]. The total synthesis was commenced from D-ribose 204, which was converted to alcohol 205 in a few steps. Then, the Mitsunobu reaction was performed to couple alcohol 205 and acid 206 in the presence of triphenyl phosphine (PPh3), diethyl azodicarboxylate (DEAD), and toluene at room temperature for 6 h to obtain esterification product 207 in an 89% yield. Further, Hoveyda Grubbs second generation catalyst was used to perform ring closing metathesis that furnished cyclized trans-macrolactone product 208 in an 80% yield. In the last step, paecilomycin E 209 was obtained in an 82% yield by employing the global deprotection of macrolactone in the presence of CH2Cl2 and BCl3 (Scheme 12Scheme 29).
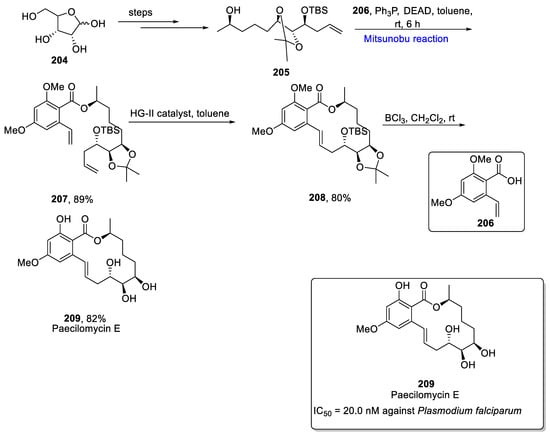
Scheme 129.
Total synthesis of paecilomycin E
209
.
Styryllactones are naturally occurring organic compounds which are isolated from a number of natural sources, such as Goniothalamus dolichocarpus and Cryptocarya caloneura [35][67]. Since these compounds are biologically active, they have been used in Thai traditional medicine and exhibit anti-cancerous activity [36][68]. Kotammagari et al. proposed the total synthesis of goniothalamin and its epimers by using Ferrier reaction, Jones oxidation, acid-mediated transition metal free epimerization, and Mitsunobu reaction as key steps, in 2019 [37][69]. The total synthesis was commenced from triacetyl-O-D-glucal 210, which was converted to compound 211 over a few steps. Next, 85% of alcohol 212 was obtained, which underwent a Mitsunobu inversion to invert stereochemistry of the OH group in the presence of p-nitrobenzoic acid (PNBA) and diethyl azodicarboxylate (DIAD) at 0 °C to room temperature for 12 h, followed by deprotection of the ester to obtain 60% of epimerized alcohol 213. Further, the epimerized alcohol 213 was protected in the presence of tert-butyl dimethyl-silyl chloride (TBSCl) and imidazole followed by Jones oxidation and deprotection of the TBS group in the presence of BF3.OEt2 to furnish the final compound (−)-5-hydroxygoniothalamin 214 in a 30% yield along with its C-5 epimer. The compound 211 was converted to 62% of (+)-5-hydroxygoniothalamin and its C-5 epimer by oxidation in the presence of Jones reagent and deprotection in the presence of BF3.OEt2. The synthesis of (−)-5-acetylgoniothalamin 216 was accomplished by the acetylation of (−)-5-hydroxygoniothalamin 214 in an 85% yield (Scheme 13Scheme 30).
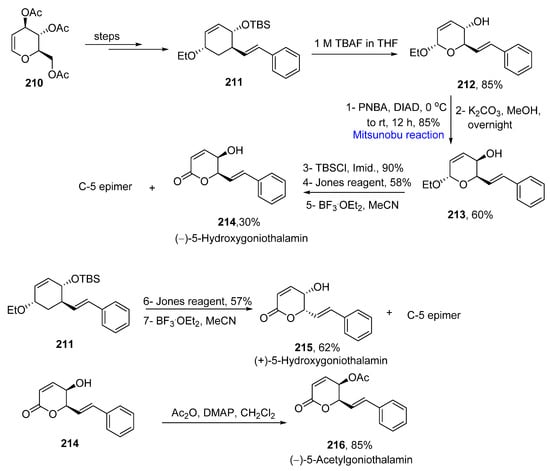
Scheme 130.
Total synthesis of (−)-5-hydroxy goniothalamin
214
, (+)-5-hydroxy goniothalamin
215
and (−)-5-acetylgoniothalamin
216
.
Limaol is a cytotoxic agent extracted from a marine dinoflagellate, P. lima [38][70]. Hess et al. reported the total synthesis of limaol in 2021 [39][71]. Stille reaction, asymmetric propargylation, a gold catalyzed-spirocyclization, Mitsunobu reaction, and substrate-controlled allylation were employed as key steps for the synthesis of limaol 223. The synthesis commenced with the preparation of fragments 219 and 220 from commercially available starting substances, such as 217, which were prepared from 217 and 218 over a few steps. Fragment 219 and 220 were made to couple together in the presence of MgBr2.OEt2 and dichloromethane to yield 88% of the coupled product 221. In the next step, the Mitsunobu reaction was performed in the presence of 4-nitrobenzoic acid, triphenyl phosphine, diethyl azodicarboxylate, and toluene at 0 °C to room temperature to invert the configuration of the OH group at C-27 in compound 222 with a yield of 91%. Finally, compound 222 was converted to limaol 223 with a yield of 32% over a few steps (Scheme 14Scheme 31).
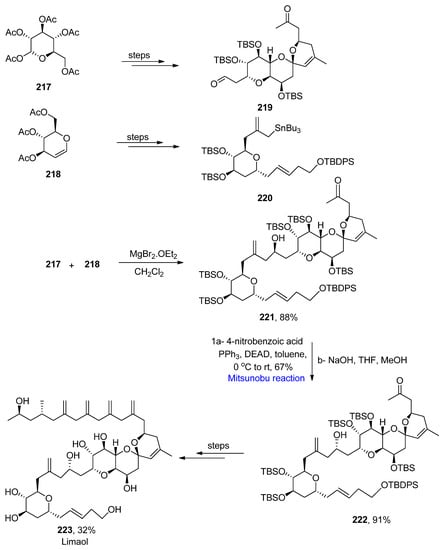
Scheme 314.
Total synthesis of limaol
223
.
Petromyroxol belongs to acetogenins, which possess cytotoxic, anti-pyrexial, anti-helminthic, and anti-cancer properties, etc. [40][72]. Petromyroxols are isolated from a larval sea lamprey and exhibit a good olfactory response. Mullapudi et al. proposed the total synthesis of (+)-petromyroxol and its diastereomers in 2020 [41][73]. For the synthesis of (+)-petromyroxol, allyl glycoside 224 was made to undergo a Mitsunobu reaction in the presence of para-nitrobenzoic acid to give unsaturated compound 225 in an 81% yield. In the next step, the compound 225 was subjected to olefin oxidative cleavage followed by two-carbon homologation to give unsaturated ester derivative 226 in a 69% yield. In the later step, hydrogenation by Pearlman catalyst along with saponification of both esters yielded (+)-petromyroxol 227 in a 77% yield (Scheme 15Scheme 32).
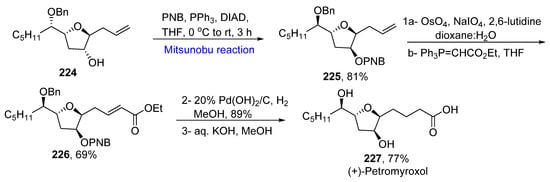
Scheme 1532.
Total synthesis of (+)-petromyroxol
227
.
For the synthesis of 6-epi-(−)-iso-petromyroxol 231, the allyl glycoside 228 was subjected to a Mitsunobu reaction in the presence of para-nitrobenzoic acid (PNB), DIAD, and PPh3 at 0 °C to room temperature for 3 h to obtain the PNB-substituted product 229 in an 86% yield. In the next step, olefin oxidation of the substituted product 228 followed by two-carbon homologation gave 71% of ester 230. The last step included Pearlman catalyst-mediated hydrogenation, followed by saponification of ester 230 to give the final product 6-epi-(−)-iso-petromyroxol 231 in a 74% yield (Scheme 15Scheme 32). Some other diastereomers of petromyroxol were also synthesized by taking an isomer of allyl glycoside 224 as starting compounds by employing a similar strategy (Scheme 16Scheme 33).
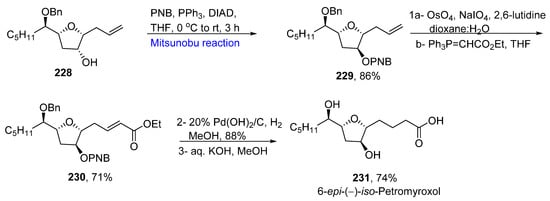
Scheme 1633.
Total synthesis of 6-
epi
-(−)-iso-petromyroxol
231
.
5. Natural Products-Based Terpenes Synthesis
Curcusones belonging to the group of diterpenes are complex natural products. They are extracted from Jatropha curcas and possess anti-cancerous activities. Cui et al. reported the first total synthesis of curcusone diterpenes in 2021 [42][74]. First of all, cyclohexenal 232 was converted to silyl enol ether, followed by a Mukaiyama aldol reaction and NaBH4 reduction to give 233 in a 73% yield. Further, an organocatalytic Mitsunobu reaction was performed in the presence of diethyl azodicarboxylate and alcohol 234 at 0 °C to 50 °C to yield compound 235. The compound 235 was converted to the enone 236 over a few steps. Next, the enone 236 was methylated in the presence of KHMDS and methyl iodide to obtain two isomers (−)-S-curcusone A and (−)-R-curcusone B 238, which were converted to (−)-R-curcusone C and (−)-S-curcusone D 240 by α-hydroxylation in 1:1 diastereomeric ratio. The compound 238 was converted to (+)-spirocurcusone 238 in a 60% yield and (−)-pyrocurcusone 239 in a 21% yield. The compound 240 was also converted to (−)-dimericursone 241 over a few steps (Scheme 17Scheme 34). The synthesized compounds were further evaluated for anti-proliferative activity against MCF-7 cells by using a WST-1 assay. Curcusone D 240 exhibited the best cytotoxic activity among all.
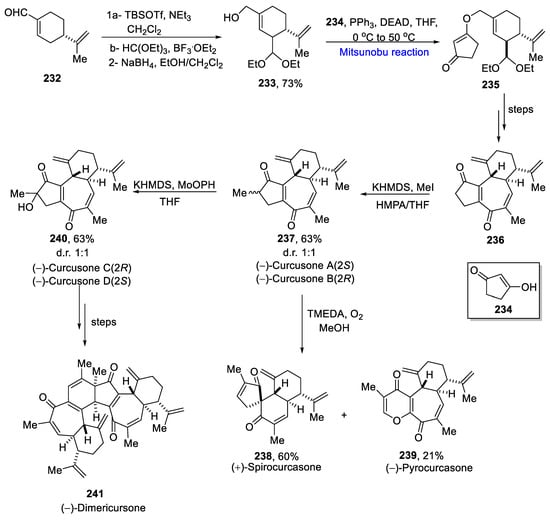
Scheme 1734.
Total synthesis of (−)-curcursone isomers
238
and
240
, and (−)-dimericursone
241
.
Vibsanins are diterpenoids present in the leaves of Viburnum awabuki, which possess piscicidal activity. They consist of 7-membered, 11-membered, and rearranged vibsanes. The total synthesis of 11-membered vibsane is reported by Takao et al. in 2020 [43][75]. The synthesis commenced from the preparation of fragments 244 and 245 from commercially available substances 242 and 243 over a few steps. These fragments were coupled in the presence of tertiary butyl and THF to give the compound 246 in a 52% yield. The Mitsunobu reaction between 246 and acid 247 at −15 °C was performed to afford acylate and give ester 248 in a 72% yield. Further, deprotection and oxidation of ester 248 followed by a Nozaki–Hiyama–Takai–Kishi (NHTK) reaction gave diastereoselectively pure alcohol 249 in an 82% yield. The last step involved another Mitsunobu reaction with para-nitrobenzoic acid, diethyl azodicarboxylate, and triphenyl phosphine at 0 °C, followed by chemoselective methanolysis to give the final product 250 in a 91% yield (Scheme 18Scheme 35).
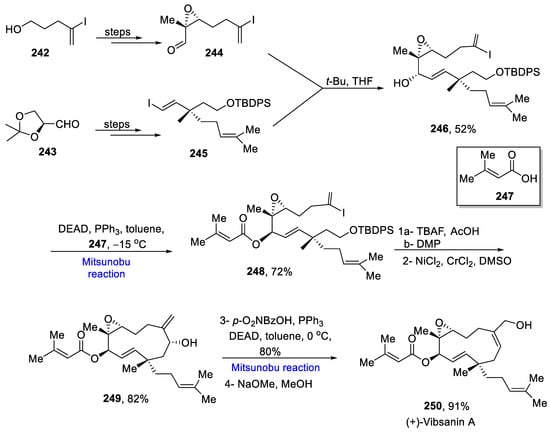
Scheme 1835.
Total synthesis of (+)-vibsanin A
250
.
Pavidolide B belongs to the cembranoid family, which is a pervasive class of marine natural products. These are isolated from genera Sarcophyton and Sinularia. These exhibit biological activity against human probe myelocytic cell lines. In 2019, Zhang et al. proposed the total synthesis of (−)- pavidolide B by utilizing annulation reactions and ring closing metathesis reactions as key steps to achieve a 16% overall yield [44][76]. The total synthesis was commenced by the reaction of unsaturated aldehyde 251 and dimethyl-2-bromomalonate 252 in the presence of 2-(diphenyl((trimethylsilyl) ocy) methyl) pyrrolidine catalyst 253 with the subsequent protection of the aldehyde group in the presence of PTSA and CH(OEt)3, which were selected after the screening of various conditions, followed by hydrolysis to give 80% of substrate 254 in a diastereomeric ratio of 1.5:1. In the later step, the Mitsunobu coupling with alcohol 255 was employed in the presence of diethoxyethyl azodicarboxylate (DEAD) to obtain 74% of the coupled product 256 in a similar diastereomeric ratio. After the screening of various conditions, the annulation reaction was performed in the presence of PhSH and AIBN to obtain the annulated product 257 in a 38% yield with a dr of 1.6:1. The compound 257 was converted to tetracyclic pavidolide 258 in a 95% yield over a few steps (Scheme 19Scheme 36).
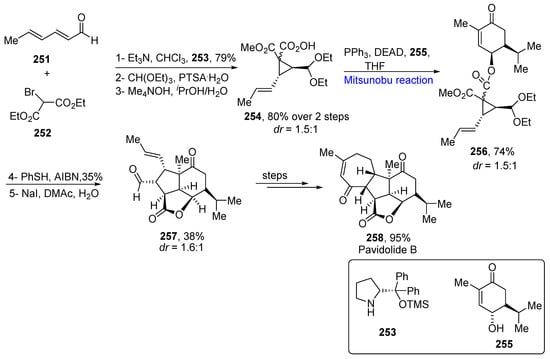
Scheme 1936.
Total synthesis of pavidolide B
258
.
(+)-ar-Macrocarpene is a naturally occurring sesquiterpenoid derived from Cupressus macrocarpa. This family covers a number of biologically active derivatives. In 2019, Khatua et al. reported the total synthesis of ar-Macrocarpene in a 42.1% overall yield from the commercially available compound 259, which was converted to alcohol 260 over a few steps [45][77]. It underwent the Mitsunobu reaction with o-nitrophenylsulfonyl hydrazide 261 at 0 °C for 2 h to furnish sulfonyl hydrazine 262, which was rearranged in the presence of methanol, and gave 65% of the diazene intermediate 263 in the presence of methanol. In the final step, the diazene intermediate was further hydrogenated to yield (+)-ar-Macrocarpene 264 in a 99% yield (Scheme 20Scheme 37).
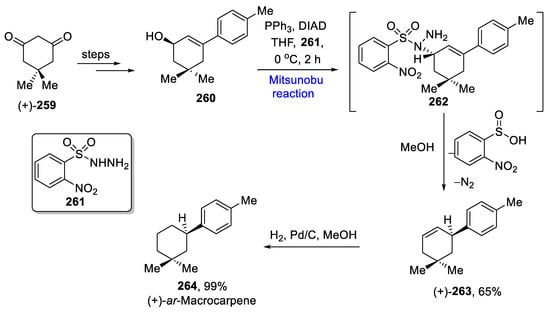
Scheme 2037.
Total synthesis of (+)-
ar
-Macrocarpene
264
.
Andrographolide is known as the king of bitters as it has a lot of applications in ayurvedic and local medicine [46][78]. Yang et al. proposed the total synthesis of andrographolide in 2020 [47][79]. Diene 265 was cyclized via Diels–Alder cycloaddition by using DMAD along with hydrolysis, followed by manganese-catalyzed HAT reduction, which gave trans-decalin 266 in a 63% yield. It was further treated with TIPSOTf and diisobutyl aluminum hydride (DIBAL-H) to produce ene-diol 267. The next step proceeded via a Mitsunobu reaction with 120 mol% of hydrazine derivatives in the presence of 120 mol% of diethyl azodicarboxylate and 130 mol% of triphenyl phosphine at −30 to 25 °C followed by the conversion to iodide 268 in the presence of imidazole and iodine in a 72% yield. In the next step, the treatment of iodide 268 with 2-thienyl copper lithium and vinyl bromide 269, along with carbonylative lactonization with Palladium-Xanthophos, gave andrographolide 270 in a 92% yield (Scheme 21Scheme 38).
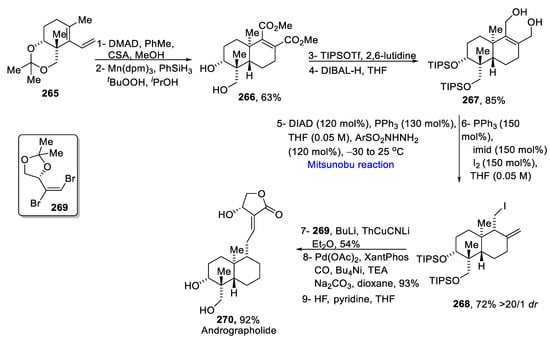
Scheme 2138.
Total synthesis of andrographolide
270
.
For the synthesis of 14-hydroxy-collandonin, diene 265 was converted to diol 271 over a few steps. The diol 271 underwent acylation that gave allylic carbonate 272 in a 74% yield [47][79]. Further, allylic carbonate 272 was reacted with 250 mol% of umbelliferone 273 via a Mitsunobu reaction in the presence of 150 mol% of diethyl azodicarboxylate and 150 mol% of triphenyl phosphine at 25 °C followed by a Tsugi reduction in the presence of palladium acetate and formic acid along with the deprotection of silyl ethers to give 14-hydroxy-colladonin 274 in a 95% yield (Scheme 22Scheme 39).
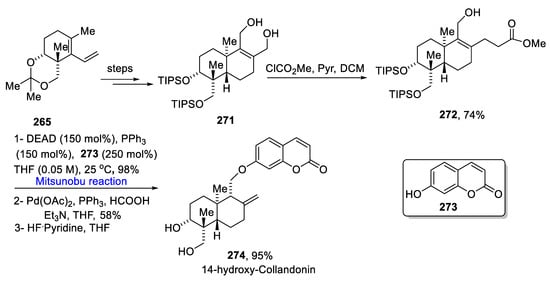
Scheme 2239.
Total synthesis of 14-hydroxy-collandonin
274
.