Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rodrigo P. Silva-Aguiar and Version 2 by Dean Liu.
O-GlcNAcylation is regulated by the enzymatic balance between O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) which add and remove GlcNAc residues on target proteins, respectively. This post-translational modification is essential for cellular physiology, and unbalanced protein O-GlcNAcylation is associated with several diseases. Here, we discuss aspects of protein O-GlcNAcylation in renal physiology and pathophysiology
- O-GlcNAcylation
- O-GlcNAc transferase
- O-GlcNAcase
- O-GlcNAc
1. O-GlcNAcylation
Initially, it was believed that covalent modifications of proteins with carbohydrates would occur only at the endoplasmic reticulum and the Golgi apparatus in proteins that are secreted or expressed at the plasma membrane in a process called N-glycosylation [1][2][28,29]. This paradigm was changed since the first description of intracellular O-GlcNAcylation outside of these compartments [3][21]. The molecular mechanisms governing O-GlcNAc homeostasis and its participation in physiology and in pathophysiology have been extensively studied and validated [4][5][6][7][8][9][10][11][22,23,24,25,26,27,30,31]. Two enzymes were described to be responsible for O-GlcNAc cycling: (1) the O-GlcNAc transferase, OGT, encoded by the gene OGT, responsible for adding O-GlcNAc moiety into serine or threonine residues of target proteins; and (2) the glycosidase O-GlcNAcase, OGA, encoded by the gene OGA, responsible for removing O-GlcNAc residues from proteins [10][30]. Both OGT and OGA are evolutionarily conserved [12][32], ubiquitously expressed in mammalian cells, and their structures have been resolved [13][14][33,34]. Deletion of both OGT and OGA is lethal in different animal models, suggesting that O-GlcNAcylation is essential for organism development [15][16][17][35,36,37].
In addition to OGA and OGT balance, O-GlcNAcylation depends on substrate availability of uridine-diphosphate-N-acetyl-glucosamine, UDP-GlcNAc [18][19][38,39]. This substrate is the donor of GlcNAc for O-GlcNAcylation and is produced by the hexosamine biosynthetic pathway (HBP) [20][40] (Figure 1). This anabolic pathway shifts glucose metabolism to produce high energy sugar intermediates for different types of glycosylation. Accordingly, pharmacological or genetic decrease in HBP flux through inhibition of HBP rate-limiting enzyme glutamine-fructose amino transferase (GFAT, expressed in two isoforms, GFAT1 and GFAT2, encoded by the genes GFPT1 and GFPT2, respectively) significantly lowers O-GlcNAcylation in vitro and in vivo [21][22][23][24][41,42,43,44]. Increases in UDP-GlcNAc levels, i.e., bypassing GFAT activity by adding glucosamine, enhances O-GlcNAcylation in vitro and in vivo [25][26][27][45,46,47]. UDP-GlcNAc is considered a central metabolic sensor because it requires inputs from nucleotide, carbohydrate, fatty acid, and amino acid metabolism [19][28][39,48].
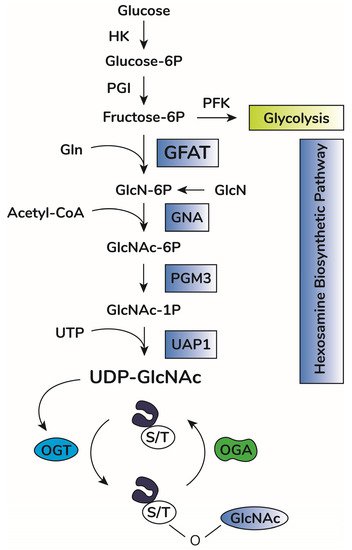
Figure 1. O-GlcNAc cycling scheme. Upon entry in cells, glucose is rapidly phosphorylated to glucose-6-phosphate (Glucose-6P) by hexokinase (HK). Glucose-6P is isomerized by phosphoglucose isomerase (PGI), producing fructose-6-phosphate (Fructose-6P), a substrate for both phosphofructokinase (PFK) of the glycolytic pathway or glucosamine-fructose amino transferase (GFAT), the rate-limiting reaction of the hexosamine biosynthetic pathway (HBP). GFAT requires glutamine (Gln) as the amine donor for generating glucosamine-6-phosphate (GlcN-6P), which is then N-acetylated by glucosamine-6-phosphate N-acetyltransferase (GNA1), producing N-acetyl-glucosamine-6-phosphate (GlcNAc-6P). This step requires acetyl-CoA as the acetyl donor. GlcNAc-6P is transformed in GlcNAc-1P by phosphoacetylglucosamine mutase (PGM3). Using UTP as the nucleotide donor, UDP-N-acetylglucosamine pyrophosphorylase (UAP1) produces uridine-diphosphate N-acetyl glucosamine (UDP-GlcNAc). This molecule is the substrate of O-GlcNAc transferase (OGT) for protein O-GlcNAcylation by adding O-linked GlcNAc moieties at serine or threonine residues of target proteins. O-GlcNAcase (OGA) removes GlcNAc residues, counteracting OGT activity.
Indeed, O-GlcNAcylation regulates several cell processes, including proliferation [29][49], differentiation [30][50], motility [31][51], and gene transcription [32][52]. This task is performed by the direct effects of O-GlcNAcylation [10][11][30,31], by noncatalytic functions of OGT [33][53], or by intricate crosstalk with other PTMs, such as phosphorylation [34][35][54,55] and ubiquitination [36][56]. At this point, an important gap is still being discussed: How is O-GlcNAcylation so abundant and diverse (i.e., it has been found in over 5000 proteins to be modified [37][38][57,58]) when cells have only one transferase and one glycosidase? Interactions between OGT or OGA with binding partners could promote selective O-GlcNAcylation of target proteins in a specific cell context [39][40][41][59,60,61]. How this is performed, however, requires further examination.
O-GlcNAc homeostasis operates under a physiological threshold of changes in cellular O-GlcNAcylation levels [10][30]. Changes beyond this threshold for excessive periods of time, however, would generate pathophysiological processes. This model fits with data observed in diabetes [42][43][44][62,63,64], cancer [45][46][47][65,66,67], and cardiovascular [26][48][49][50][46,68,69,70] and neurodegenerative diseases [51][52][53][54][71,72,73,74]. In diabetes, the hyperglycemic milieu has been shown to shift cell metabolism towards increased HBP, increasing UDP-GlcNAc production and enhancing protein O-GlcNAcylation [55][56][75,76]. This mechanism mediates hyperglycemia-induced cytotoxicity [56][76]. In cancer cells, increased proliferation is supported by metabolic adaptions such as enhanced glucose [57][77] and glutamine [58][78] uptake and enhanced ATP production through glycolysis, a condition termed Warburg effect [59][79]. Importantly, HBP-mediated UDP-GlcNAc production sustains tumor development through aberrant glycosylation and protein O-GlcNAcylation [60][61][80,81]. Acute increases in O-GlcNAcylation protect the brain against stroke [51][71] and ameliorate cognitive decline in elder mice [54][74]. This protective effect of acute elevations in O-GlcNAcylation is also seen in trauma-induced cardiac dysfunction [26][46]. Chronic changes in O-GlcNAcylation levels, however, are associated with cardiovascular diseases, such as diabetes-induced arrhythmia [50][70] and heart failure [62][82], and the development of neurodegenerative diseases, including Alzheimer’s [53][73]. This model also applies to renal physiology and pathophysiology, as discussed below.
2. O-GlcNAcylation in Renal Physiology
Transcriptomic and proteomic analysis of different nephron segments have demonstrated that OGT, OGA, and GFAT expression are higher at medullary segments, including the thin descending limb (TDL), thin and thick ascending limb of the loop of Henle, and collecting duct [63][64][83,84]. Cortical segments, including proximal tubules and distal tubules, have lower but significant expression levels. Advances in tissue-specific gene editing tools have been used to study the importance of O-GlcNAcylation in specific extrarenal cells [65][66][67][68][69][70][85,86,87,88,89,90]. Ono et al. (2017) [71][91] accessed the importance of O-GlcNAcylation for podocytes, differentiated epithelial cells involved in selective permeability through the glomerular filtration barrier [72][92]. Congenital deletion of OGT in podocytes caused progressive podocyte loss, albuminuria, glomerulosclerosis, and subsequent tubule-interstitial injury. This was associated with reduced podocin expression, podocyte effacement, and disruption of slit diaphragm structure. Interestingly, tamoxifen-induced OGT deletion in adult mice did not cause significant changes in albuminuria or podocyte structure [71][91]. Thus, OGT and, consequently, O-GlcNAcylation appear to be essential for podocyte development, but not necessary for differentiated podocyte function in physiological conditions.
Sugahara and colleagues (2019) [73][93], using a tamoxifen-induced, proximal tubule-specific OGT deletion in adult mice, did not observe changes in renal function or urine excretion of different solutes reabsorbed by PTECs (proximal tubular epithelial cells) under ad libitum-fed conditions. However, in fasting conditions, OGT deletion caused PTEC apoptosis associated with widespread reduction in reabsorption capacity, reflected by urinary loss of glucose, albumin, amino acids, phosphate, calcium, and other solutes [73][93]. This was not associated with significant changes in the mRNA expression of different transporters such as SGLT2 and NaPiIIa, involved in the reabsorption of glucose and phosphate, respectively. Rather, significant metabolic changes were observed. Absence of OGT decreased PTEC lipolysis, the principal energy source of PTECs in fasting conditions. Furthermore, OGT KO exacerbated tubule interstitial injury induced by a high fat diet [73][93]. These results are in line with previous reports that changes in PTECs metabolism is associated with renal damage [74][94].
So far, the results available in the literature point to a role for OGT in early glomerulus development and maintenance of physiological proximal tubule metabolism, contributing to proper reabsorption capacity in this segment [71][73][91,93]. Future directions should include the use of OGA deletion models for understanding the impact of increased O-GlcNAcylation for renal physiology, as well as assessing the influence of O-GlcNAcylation in different renal cell types.