Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Takeshi Yamauchi.
Malignant melanoma is an aggressive cancer of the skin and the leading cause of death from skin cancer. One major risk factor linked to melanoma development is exposure to UV radiation. Recent studies have demonstrated that alcohol consumption is positively linked with an increased risk of cancers, including melanoma.
- melanoma
- ethanol metabolism
- alcohol
- ethanol
- acetaldehyde
- alcohol dehydrogenase
- aldehyde dehydrogenase
1. Introduction
Cutaneous melanoma is an aggressive malignancy of the skin and a significant public health concern. Melanoma incidence continues to rise globally, placing a greater burden on industrialized countries [1,2,3,4,5][1][2][3][4][5]. In the United States, melanoma remains the fifth most common cancer, making up 6 and 5 percent of all new cancers in males and females, respectively [6,7][6][7]. Although melanoma mortality decreased from 2017 [8] to 2021 [9], the incidence continues to increase: approximately 99,780 new melanoma cases will be diagnosed in the United States in 2022 [6], almost doubling from 53,600 in 2002 [10]. The increased incidence of cutaneous melanoma has been hypothesized to be, in part, the result of improved screening and early detection. However, the incidence of advanced melanoma is also increasing, suggesting that risk factors for melanoma are on the rise [11,12,13][11][12][13].
Risk factors and conditions for the development of melanoma are categorized into three: (1) genetic risk factors, (2) phenotypic risk factors reflecting gene/environment interactions, and (3) social-environmental risk factors [14]. Genetic risk factors include family history, light skin/hair/eye color, DNA repair defects, and several melanoma risk genes, such as cyclin-dependent kinase (CDK) inhibitor 2A (CDKN2A), CDK4, BRCA1-associated protein-1 (BAP1), protection of telomeres 1 (POT1), and telomerase reverse transcriptase (TERT) [15,16][15][16]. Mutations in these tumor suppressor genes confer high susceptibility to melanoma. In contrast, some genetic factors, especially when interacting with phenotypic and environmental risk factors, have great significance in melanoma susceptibility. For example, melanocortin 1 receptor (MC1R) R (D84E, R142H, R151C, I155T, R160W, D294H) variants are associated with the fair skin and red hair color phenotype, which is prone to sunburn and has an increased risk of melanoma [15,16][15][16]. Phenotypic expressions of gene/environment interactions also include a personal history of skin cancer and numerous nevi [17,18,19,20,21,22][17][18][19][20][21][22]. While identifying these high-risk cohorts improves public health and clinical care management, genetic factors are unchangeable. Therefore, recognizing modifiable risk factors, such as social-environmental risk factors, is crucial from the clinical perspectives of patient outreach, education, and disease management.
The most notable social-environmental risk factors include ultraviolet (UV) exposure, tanning bed use, pregnancy, and chemical or carcinogen exposure [23,24,25,26][23][24][25][26]. Due to its strong genotoxic effects, melanoma development has been most commonly linked with UV radiation. Mutations caused by UV radiation account for >70% of the nucleotide mutations found in melanoma cases [27,28][27][28]. However, not all changes involved in melanoma incidence are UV-induced [29], and melanoma does not always occur in sun-exposed areas, a key difference from non-melanoma skin cancer [30,31][30][31]. Nearly one-third of melanoma cases are present in areas of the skin not usually exposed to UV light [31]. A small percentage of melanoma also occurs on mucosal surfaces, typically not exposed to the sun, and they tend to have a worse prognosis [32]. These findings suggest that UV exposure alone does not explain the sharply increased incidence of melanoma [7].
Pregnancy has been considered a trigger for melanoma since the 1950s [33,34][33][34]. A study of 1,309,501 maternities aged 15–44 years from 1994–2008 in New South Wales, Australia, found that the ratio of age-adjusted observed-to-expected rates for melanoma was 2.22 (95% CI = 2.05–2.41) [35,36][35][36]. While approximately one-third of melanoma cases in women are diagnosed during their childbearing age [37], these populations represent about 15% of melanoma cases. As melanoma incidence has increased similarly between males and females (1.90- and 1.81-fold increase from 2002 to 2022, respectively) [6[6][10],10], pregnancy alone does not explain the continued rise in melanoma cases. Other social-environmental risk factors such as older patients, organ transplant patients, and those with a history of immunosuppressive therapy are also conditions that have shown a significant correlation with aggressive melanoma, greater incidence of metastases, and lower survival rates [38]. These factors, however, are not modifiable.
Recent studies have shown links between melanoma incidence and other modifiable social-environmental factors such as obesity, tobacco use, and alcohol consumption. Obesity negatively impacts outcomes for surgically resected melanoma but leads to better outcomes when treated with immunotherapy [39,40][39][40]. However, the International Agency for Research on Cancer (IARC) found no evidence to correlate obesity and melanoma after reviewing more than 1000 epidemiological studies [41]. Linking tobacco use and increased risk of skin malignancies, especially melanoma, has been a topic of great investigation. However, the data remains unclear. While the IARC has declared smoking a cause of 18 cancers, cutaneous malignancies are not included in this data [42]. On the other hand, many studies have found a positive correlation between alcohol consumption and increased melanoma incidence.
2. Potential Roles of Ethanol on Melanoma Initiation and Progression
Absorption of orally administered alcohol beverages depends on ethanol concentration, blood flow, rate of ingestion and gastric emptying, beverage type, food intake, and the irritant properties of ethanol [84][43]. When ingested, ethanol is oxidized to toxic acetaldehyde (AcAH) by alcohol dehydrogenase (ADH) and then to acetic acid by mitochondrial aldehyde dehydrogenase 2 (ALDH2) (Figure 21, ethanol to a right direction) [84][43].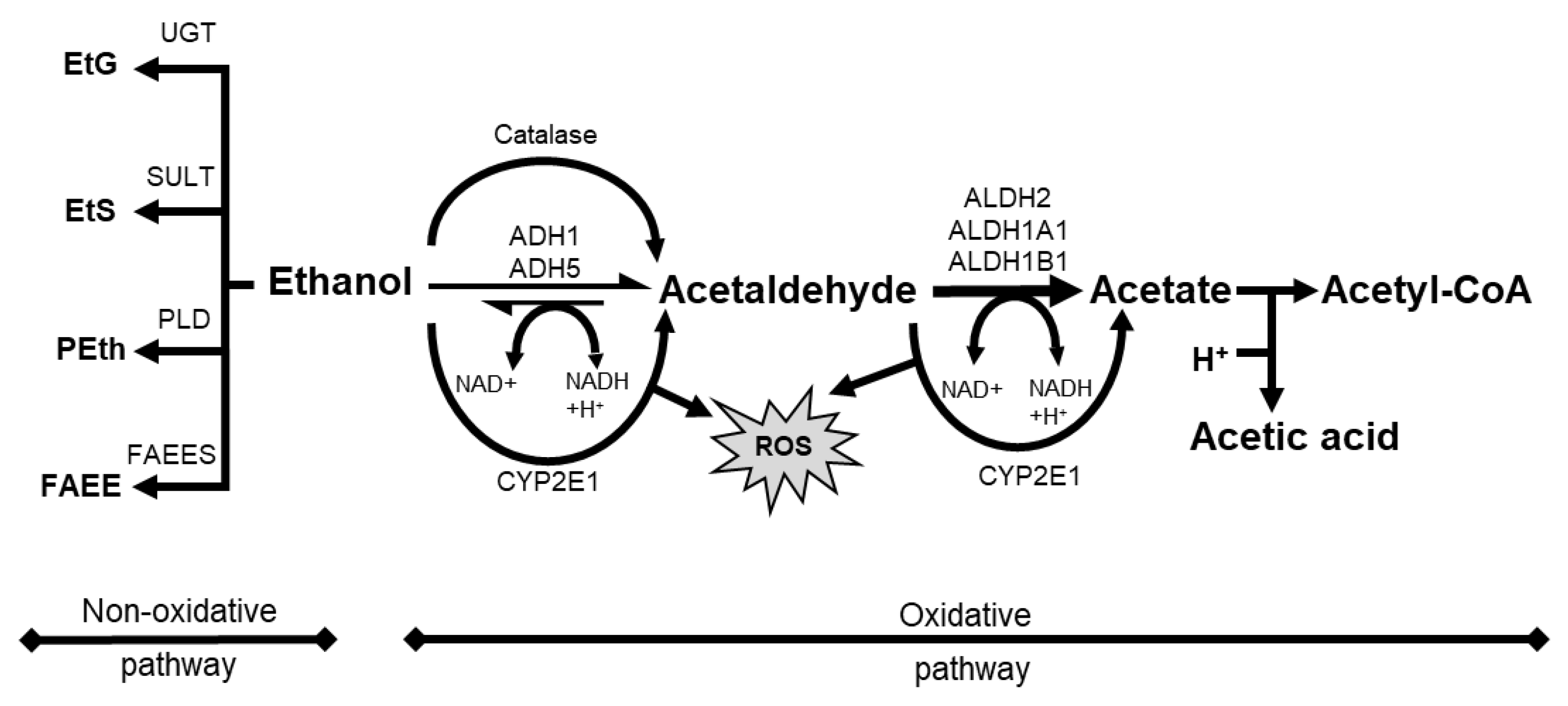
Figure 21. Schematic overview of alcohol metabolism with oxidative pathway and non-oxidative pathway. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CYP2E1, cytochrome P 450 2E1; EtS, ethyl sulfate; FAEE, fatty acid ethyl ester; FAEES, fatty acid ethyl ester synthase; NAD, nicotinamide adenine dinucleotide; Peth, phosphatidyl ethanol; PLD, phospholipase D; ROS, reactive oxygen species; SULT, sulfotransferase; UGT, UDP-glucuronosyltransferase.
2.1. Roles of Ethanol or AcAH in Cellular Biology
Ethanol and/or AcAH induce oxidative stress, DNA damage, and lipid peroxidation, which activate protein kinases and signaling pathways implicated in glycolysis, fatty acid oxidation, inflammation, differentiation, angiogenesis, and metastasis, thereby creating a favorable microenvironment for tumor initiation and progression [94][56]. Both ethanol and AcAH promote oxidative stress. After ethanol uptake, ADH-catalyzed reactions in the cytosol and ALDH2-mediated reactions in the mitochondria reduce an NAD+/NADH redox ratio [84][43], regenerating NAD+ from NADH via the mitochondrial electron transfer system with concomitant ROS production [94][56]. AcAH is a highly reactive metabolite and a mutagen. AcAH-mediated DNA damage includes adduct formation, double-strand breaks, point mutations, DNA-DNA cross-links, sister chromatid exchanges, and chromosomal aberrations [95][57]. AcAH binds proteins involved in DNA repair and methylation, altering their structure and functions and promoting carcinogenesis [96,97,98][58][59][60]. AcAH also reacts with deoxyguanosine residues, leading to DNA modifications and lesions [99][61], impairing replication, transcription, and metabolism, and increasing mutation rates and cell death [99][61]. Another mutagenic effect of ethanol and/or AcAH is mediated by CYP2E1 induction, resulting in increased ROS generation. ROS-induced lipid peroxidation products such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are genotoxic, thus generating mutagenic DNA adducts [100][62]. In addition, following ethanol intake, hybrid adducts can be generated in the affected tissues, such as the hybrid MDA and AcAH-protein adducts, increasing the tumorigenic potential of individual adducts by reducing NAD+ to NADH [100,101][62][63].2.2. Roles of Ethanol or AcAH in Tumor Biology
The abovementioned changes activate multiple signal transduction mechanisms, such as cAMP/PKA signaling [102[64][65][66],103,104], mitogen-activated protein kinase (MAPK) signaling [105][67], PI3K/Akt signaling [106[68][69],107], and Wnt/β-catenin signaling [108][70] (Figure 32). For example, ethanol stimulates cAMP-mediated PKA activation [104[66][71][72],109,110], and PKA activation has tumor-promoting or tumor-suppressive effects [111,112][73][74]. Ethanol also activates PKC [113][75]. Activated PKC induces RAS, RAF, or MEK 1/2 activation, leading to the activation of MAPK signaling to proliferate mammalian cells [114][76] and PI3K/Akt signaling to regulate cell survival and proliferation [115][77]. Furthermore, chronic alcohol consumption upregulates Wnt/β-catenin signaling, leading to tumor formation and progression in the liver cancer model [108][70] and tumor invasion in the colon cancer model [116][78]. Ethanol also induces JNK1-dependent upregulation of Brf1 expression and RNA Pol III gene transcription in breast cancer [117][79] and ethanol-induced liver cancer [118][80]. In addition, ethanol-induced genotoxic stress and oxidative stress can activate p53, which in turn activates sphingolipid-metabolizing enzymes, resulting in the accumulation of the ceramide metabolite sphingosine-1-phosphate (S1P), a promoter of the proliferation and inflammation in carcinogenesis [119][81]. These signaling pathways are key in initiating cellular responses implicated in tumorigenesis and progression, such as proliferation, differentiation, development, inflammation, survival, and cell death.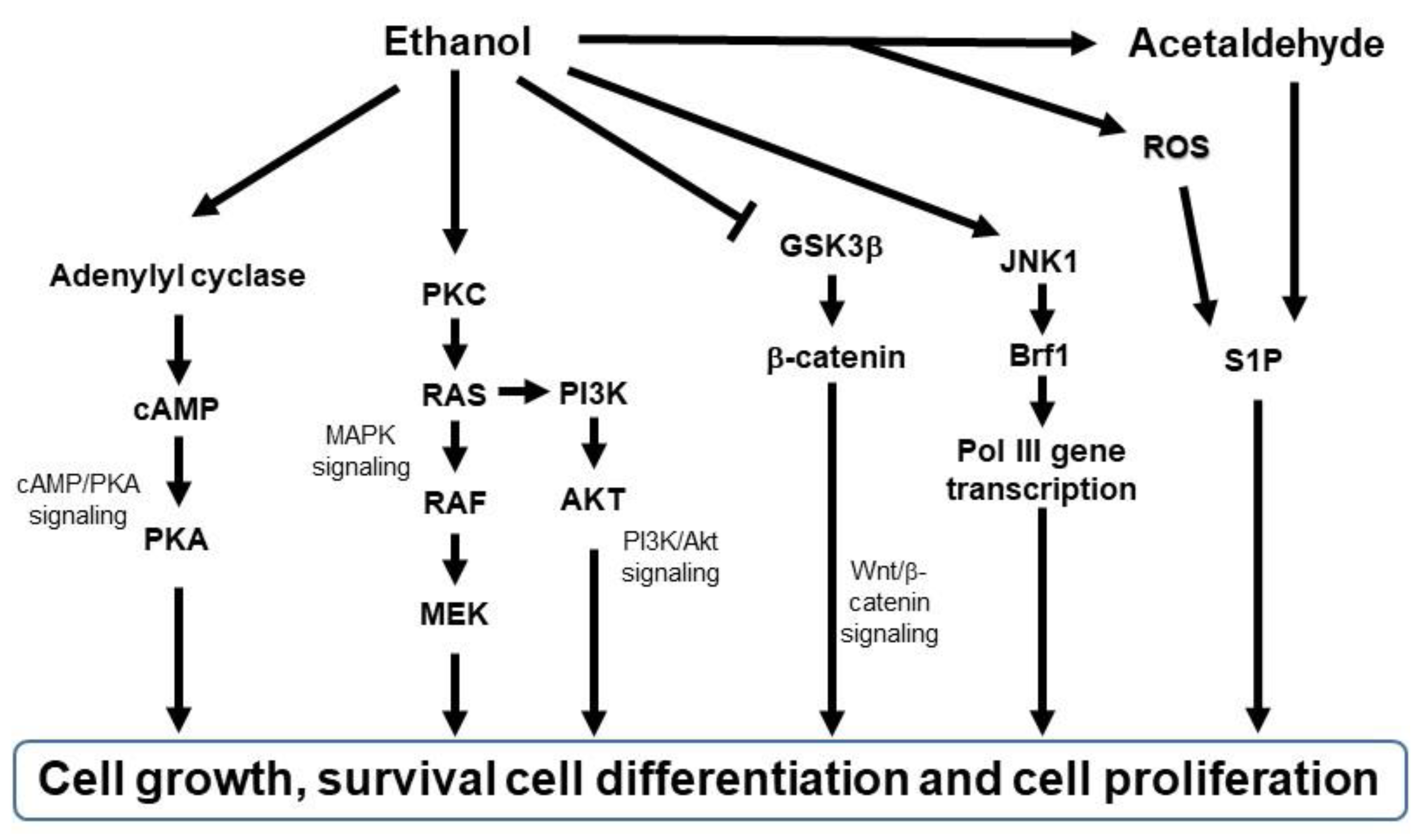
Figure 32. Schematic diagram of signaling pathways activated by ethanol. Brf1, TFIIB-related factor 1; AKT, protein kinase B; cAMP, cyclic adenosine monophosphate; GSK3β, glycogen synthase kinase 3 beta; JNK1, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK/Erk kinase; PI3K, phosphatidylinositol-3-kinase; PKA, protein kinase A; PKC, protein kinase C; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma virus; ROS, reactive oxygen species; S1P; sphingosine-1-phosphatase.
2.3. Roles of Ethanol or AcAH in Skin Biology
Most research on alcohol consumption is centered around its effects on the liver and gastrointestinal tract but rarely on the skin [122,123][84][85]. Nonetheless, acute and chronic alcohol consumption induces various skin changes. Alcohol flush reaction is a typical acute response in which the rapid elevation of AcAH in the blood after drinking alcoholic beverages leads to erythema on the face, neck, and even the entire body [124][86], and this reaction occurs not only in ALDH2-deficient Asians but also in Caucasians and Native Americans [125,126][87][88]. Chronic alcohol consumption induces many skin changes such as jaundice, hyperpigmentation, and telangiectasis, which are often considered clinical manifestations of hepatic and vascular consequences [127,128][89][90]. However, it is crucial to consider ethanol’s direct effects on skin cells. For a long time, it has been known that ingested ethanol is secreted by the eccrine glands of human skin [129[91][92],130], with an almost equal concentration to blood concentration [131][93]. Ethanol can directly influence skin structure by disrupting skin cell membranes that form an effective barrier [132][94]. Furthermore, many skin microbiotas such as Cutibacterium acnes, Staphylococcus aureus, and Staphylococcus epidermidis possess ADH to convert ethanol to AcAH [133][95]. Therefore, it is likely that ethanol and AcAH exposure in the skin affects skin cell biology. The fact that chronic alcohol consumption causes esophageal melanosis in alcoholics [134,135,136][96][97][98] and skin hyperpigmentation in the epidermis of the paw and tail in mice [137,138,139][99][100][101] suggests that chronic alcohol ingestion promotes melanocyte changes. In addition to oxidative metabolism, a smaller fraction of ethanol undergoes a non-oxidative route of metabolism (Figure 21, ethanol to a left direction). It results in the enzymatic conjugation of ethanol to endogenous metabolites, yielding ethyl glucuronide (EtG), ethyl sulfate (EtS), phosphatidylethanol (PEth), and fatty acid ethyl esters (FAEE) [140][102]. While only a minor fraction of total ethanol undergoes these metabolic pathways, the resulting metabolites such as EtG remain in the blood, urine, and hair for a long time (up to several months in hair). Therefore, these biometabolites are suitable biomarkers for recent alcohol use and abuse in clinical and forensic settings [141,142][103][104]. EtG and EtS are involved in toll-like receptor signaling, oxidative stress, and lower energy metabolism [143][105]. In contrast, PEth and FAEE interfere with cellular signaling pathways and disrupt organelle function [140][102]. Therefore, these biometabolites can also contribute to direct ethanol toxicity in organs with a limited oxidative capacity [140][102]. Furthermore, chronic alcohol consumption impairs skin immunology directly and/or indirectly by altering skin Langerhans cells [144[106][107],145], migrating dendritic cells [146][108], and multiple skin T cells [146][108]. Due to ourthe use of alcohol-containing products and alcoholic beverages, these direct and indirect effects of ethanol and AcAH on ourhuman skin may not be easily eliminated. Together with the impact of skin microorganisms and potentially synergistic influences from sun exposure, these diverse effects on ourhuman skin may likely contribute to activating and transforming skin cells.2.4. Does Ethanol or AcAH Affect Melanoma Initiation?
While mutagenic effects of ethanol and/or AcAH have been demonstrated in other cancers, their contribution to melanoma remains largely elusive. Alcohol consumption lowers carotenoid levels in the plasma [147][109]. Carotenoids such as beta-carotene or lycopene can act as anti-oxidants to scavenge singlet molecular oxygen and peroxyl radicals generated during photo-oxidation and reduce solar light simulator-induced erythema [148][110]. AcAH is also a highly reactive chemical that serves as a photosensitizer [149][111]. Therefore, Darvin et al. hypothesized that alcohol consumption increased photosensitivity in human skin and recruited six male Caucasian volunteers [150][112]. They reported a decrease in the skin carotenoid concentration and minimal erythema dose (MED) after consuming 1 mL of ethanol/kg of body weight (corresponding to ~150 mL of vodka). However, these decreases were not observed after a combined intake of alcohol and ~1 liter of orange juice, rich in carotenoids. Low carotenoid levels increase erythema following UV exposure [148[110][113][114],151,152], and carotenoid consumption in the diet has been associated with decreased melanoma risk [59][115]. Therefore, these data suggest that alcohol consumption is associated with increased melanoma risk by lowering carotenoid levels and increasing UV sensitivity, indicating the synergistic effects of ethanol with UV exposure. To elucidate the synergistic effects of ethanol and UV exposure on skin cells, Brand et al. used mouse models and human skin explants [153][116]. They demonstrated that combined ethanol consumption and UV exposure increased immune dysfunction and skin damage by decreasing DNA repair capacity and inhibiting protective mechanisms such as melanin production and anti-oxidants against UV exposure. However, the mechanisms of melanoma development induced by UV light and ethanol are unclear. As mentioned in Section 3.1, excessive ethanol induces a complicated cellular response, from oxidative stress and persistent inflammation to mitochondrial DNA damage and signaling pathway activation [154[117][118],155], all implicated in tumor development. Among these pathways, MAPK signaling is one of the major pathways activated by mutations and is critical for melanoma initiation [156][119]. Strickland et al. reported that treating C3H/HeNCr mice with UV light and topical ethanol application (25% in water) thrice weekly for about 30 weeks induced primary cutaneous melanoma in 20 to 30% of the mice [157][120]. The frequency of melanoma induction was similar to that of squamous cell carcinoma. Topical ethanol application alone did not induce melanoma, and UV alone rarely induced melanoma. Interestingly, these melanoma tumors possessed Nras mutations at codons 13 and 19 in both tumors [158][121], which occurred at pyrimidine dimer sites, exhibiting a C to T transition on the non-transcribed strand at codon 13 and transcribed strand at codon 19, implicating UV-associated changes [159][122]. BRAF and NRAS are two major genes often mutated in human melanoma and are associated with melanoma initiation and progression [160][123]. Active NRAS mutations induce both MAPK and PI3K/Akt signaling [161][124]. Furthermore, these two mouse tumors and the cell lines had either a deletion in exon 2 of the Ink4a/Arf gene or an interstitial deletion of the long arm of chromosome 4 (where the Ink4a/Arf gene resides), similar to genetic changes of human melanoma for CDKN2A, encoding p16INK4a and p19ARF [162][125]. While UV is more frequently associated with the development of non-melanoma skin cancer than melanoma, it is unclear how the combination of ethanol and UV induced a relatively equal number of melanoma tumors compared to squamous cell carcinoma [157][120]. As ethanol stimulates cAMP-mediated PKA activation [104[66][71][72],109,110], this signaling may rewire β-catenin to activate the transcription of CREB target genes, including microphthalmia-associated transcriptional factor (MITF) [163][126], a master regulator of melanocyte biology [164,165][127][128]. Alterations in the MITF gene and pathway are associated with a higher risk of melanoma initiation [166,167][129][130]. MITF is regulated by several other transcription factors, including SOX10, CREB, Pax3, Tyro3, and TCF/LEF. The activation of the BRAF V600E/ERK pathway can also enhance the expression of MITF by directly phosphorylating MITF at Ser73 or activating CREB through MSK [164,168,169,170][127][131][132][133]. The PI3K/Akt signaling pathway regulates MITF through inactivating GSK-3β or cooperating with RAS/RAF/MEK/ERK signaling. In addition, MITF is a target of the p38/MAPK signaling but can be inhibited by the JNK/MAPK pathway, suggesting that the regulation of MITF is accomplished by various MAPK signaling pathways [171,172][134][135]. Considering these data collectively, it is likely that such signaling pathways are critical components of ethanol-induced carcinogenesis in some cancers, including melanoma.2.5. Does Ethanol or AcAH Affect Melanoma Progression?
Several studies have reported the biological effects of chronic alcohol consumption on melanoma progression and metastasis. Tan et al. found that ethanol-treated B16F10 melanoma tumors exhibited enhanced angiogenesis through increased vascular endothelial growth factor (VEGF) expression, contributing to tumor progression [173][136]. On the other hand, Meadow’s research team from Washington State University studied alcohol’s impact on tumor metastasis in C57BL/6 mice using B16BL6 melanoma, a derivative of B16F10 with a more invasive and metastatic phenotype [174,175,176,177][137][138][139][140]. Melanoma cells were injected subcutaneously to assess spontaneous metastasis and intravenously to assess experimental metastasis. While pretreating tumor cells in vitro or in vivo with ethanol enhanced experimental metastasis [174][137], pretreating mice with ethanol (10–20% (w/v) ethanol for >4 weeks) inhibited spontaneous and experimental metastases [174,177][137][140]. These data suggest that ethanol directly potentiates the metastatic capacity of melanoma cells. They also indicate that the host environment at the tumor injection determines the ethanol’s effect on tumor metastasis. Interestingly, survival times were significantly shorter in mice pretreated with ethanol, despite having fewer metastases [174][137], implicating the detrimental effects of ethanol in tumor-bearing mice. Several mechanisms could be involved in alcohol-mediated melanoma progression and metastasis. For example, ethanol disturbs mitochondrial dynamics [178][141] by increasing mitochondrial fission and reducing their fusion [179][142], and altered mitochondrial dynamics can promote tumor migration and progression in human melanoma [180][143]. ROS from mitochondria upregulates hypoxia-inducible factor-1α, inducing matrix metalloproteinases and VEGF [181][144], important for tumor invasion. Furthermore, DNA and ATP leaked from damaged mitochondria activate inflammasomes [181[144][145],182], reshaping the tumor microenvironment and immune infiltration to support tumor progression and drug resistance, as shown in ouresearchers' previous reports [183,184,185][146][147][148]. Another consequence of chronic and acute alcohol consumption is interference with immune cell numbers and function, which may facilitate melanoma progression and metastasis [163,186][126][149]. B16BL6 melanoma-bearing mice exposed to chronic ethanol showed fewer mature B cells, CD8+ T cells, and NK cells in circulation due to ethanol-induced downregulation of S1P/S1P receptor 1 signaling resulting in decreased egress of lymphocytes from the spleen [187][150]. Chronic ethanol administration also impairs the trafficking of NK cells to lymph nodes, resulting in a decreased number and percentage of NK cells in the draining nodes [188][151]. CD8+ T cells and NK cells are important in inhibiting tumor progression. Moreover, chronic ethanol exposure in mice impairs antigen-specific response to melanoma cells by inhibiting the proliferation of memory T cells, reducing IFN-γ producing CD8+ T cells, and increasing myeloid-derived suppressor cells [189][152]. Ethanol can also upregulate the expression of nerve growth factor receptor (NGFR/CD271) through NF-κB signaling in human melanoma cells [190][153]. NGFR expression in melanoma cells is associated with increased metastasis and long-term growth [191,192][154][155]. NGFR signaling also plays a critical role in acquired melanoma resistance to BRAF/MEK inhibitors [193,194,195,196][156][157][158][159]. VEGF expression, which contributed to angiogenesis in the melanoma mice model, is also reported to induce immune resistance by affecting myeloid-derived suppressor cells, dendritic cells, T regulatory cells, and cytotoxic T cells [173,197][136][160]. In addition, NGFR expression induced by ethanol is linked to immunosuppressive functions and anti-PD-1 immunotherapy resistance in melanoma [198,199,200][161][162][163]. Together, the accumulated data suggest a link between chronic alcohol consumption and melanoma progression. Further studies are required to ascertain these findings, especially in human melanoma patients, and elucidate the underlying mechanisms and biology. In addition to the factors mentioned above, the role of other potential factors such as genetic instability, metabolic rewiring, and skin microbiota need to be determined for a comprehensive understanding of alcohol-associated melanoma progression.References
- Bell, K.J.L.; Cust, A.E. Beyond country-specific incidence and mortality: The global burden of melanoma. Br. J. Dermatol. 2018, 178, 315–316.
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and risk factors of melanoma. Surg. Clin. N. Am. 2020, 100, 1–12.
- Roush, G.C.; McKay, L.; Holford, T.R. A reversal in the long-term increase in deaths attributable to malignant melanoma. Cancer 1992, 69, 1714–1720.
- Linos, E.; Swetter, S.M.; Cockburn, M.G.; Colditz, G.A.; Clarke, C.A. Increasing burden of melanoma in the United States. J. Investig. Dermatol. 2009, 129, 1666–1674.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Welch, H.G.; Mazer, B.L.; Adamson, A.S. The rapid rise in cutaneous melanoma diagnoses. N. Engl. J. Med. 2021, 384, 72–79.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Jemal, A.; Thomas, A.; Murray, T.; Thun, M. Cancer statistics, 2002. CA Cancer J. Clin. 2002, 52, 23–47.
- Guy, G.P., Jr.; Machlin, S.R.; Ekwueme, D.U.; Yabroff, K.R. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am. J. Prev. Med. 2015, 48, 183–187.
- Herbert, A.; Koo, M.M.; Barclay, M.E.; Greenberg, D.C.; Abel, G.A.; Levell, N.J.; Lyratzopoulos, G. Stage-specific incidence trends of melanoma in an English region, 1996–2015: Longitudinal analyses of population-based data. Melanoma Res. 2020, 30, 279–285.
- Hubner, J.; Waldmann, A.; Eisemann, N.; Noftz, M.; Geller, A.C.; Weinstock, M.A.; Volkmer, B.; Greinert, R.; Breitbart, E.W.; Katalinic, A. Association between risk factors and detection of cutaneous melanoma in the setting of a population-based skin cancer screening. Eur. J. Cancer Prev. 2018, 27, 563–569.
- Bolognia, J.L.; Jorizzo, J.L.; Schaffer, J.V. Dermatology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2012.
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14.
- Hawkes, J.E.; Truong, A.; Meyer, L.J. Genetic predisposition to melanoma. Semin. Oncol. 2016, 43, 591–597.
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018, 10, 1143–1154.
- Wei, E.X.; Li, X.; Nan, H. Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J. Am. Acad. Dermatol. 2019, 81, 489–499.
- Liang, X.S.; Pfeiffer, R.M.; Wheeler, W.; Maeder, D.; Burdette, L.; Yeager, M.; Chanock, S.; Tucker, M.A.; Goldstein, A.M.; Yang, X.R. Genetic variants in DNA repair genes and the risk of cutaneous malignant melanoma in melanoma-prone families with/without CDKN2A mutations. Int. J. Cancer 2012, 130, 2062–2066.
- Bataille, V. It’s Not All Sunshine: Non-sun-related Melanoma risk-factors. Acta Derm. Venereol. 2020, 100, adv00137.
- Rigel, D.S.; Carucci, J.A. Malignant melanoma: Prevention, early detection, and treatment in the 21st century. CA Cancer J. Clin. 2000, 50, 215–236; quiz 237–240.
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. MC1R variants, melanoma and red hair color phenotype: A meta-analysis. Int. J. Cancer 2008, 122, 2753–2760.
- Belbasis, L.; Stefanaki, I.; Stratigos, A.J.; Evangelou, E. Non-genetic risk factors for cutaneous melanoma and keratinocyte skin cancers: An umbrella review of meta-analyses. J. Dermatol. Sci. 2016, 84, 330–339.
- Pion, I.A.; Rigel, D.S.; Garfinkel, L.; Silverman, M.K.; Kopf, A.W. Occupation and the risk of malignant melanoma. Cancer 1995, 75, 637–644.
- Ting, W.; Schultz, K.; Cac, N.N.; Peterson, M.; Walling, H.W. Tanning bed exposure increases the risk of malignant melanoma. Int. J. Dermatol. 2007, 46, 1253–1257.
- Rockley, P.F.; Trieff, N.; Wagner, R.F., Jr.; Tyring, S.K. Nonsunlight risk factors for malignant melanoma. Part I: Chemical agents, physical conditions, and occupation. Int. J. Dermatol. 1994, 33, 398–406.
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263.
- Sanchez, M.I.; Grichnik, J.M. Melanoma’s high C>T mutation rate: Is deamination playing a role? Exp. Dermatol. 2014, 23, 551–552.
- Okura, R.; Yoshioka, H.; Yoshioka, M.; Hiromasa, K.; Nishio, D.; Nakamura, M. Expression of AID in malignant melanoma with BRAF(V600E) mutation. Exp. Dermatol. 2014, 23, 347–348.
- Larese Filon, F.; Buric, M.; Fluehler, C. UV exposure, preventive habits, risk perception, and occupation in NMSC patients: A case-control study in Trieste (NE Italy). Photodermatol. Photoimmunol. Photomed. 2019, 35, 24–30.
- D’Orazio, J.A.; Marsch, A.; Lagrew, J.; Veith, W.B. Skin pigmentation and melanoma risk. In Advances in Malignant Melanoma—Clinical and Research Perspectives; IntechOpen: London, UK, 2011.
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nistico, S.P. Primary mucosal melanoma presenting with a unilateral nasal obstruction of the left inferior turbinate. Medicina 2021, 57, 359.
- Jhaveri, M.B.; Driscoll, M.S.; Grant-Kels, J.M. Melanoma in pregnancy. Clin. Obstet. Gynecol. 2011, 54, 537–545.
- Driscoll, M.S.; Martires, K.; Bieber, A.K.; Pomeranz, M.K.; Grant-Kels, J.M.; Stein, J.A. Pregnancy and melanoma. J. Am. Acad. Dermatol. 2016, 75, 669–678.
- Lee, Y.Y.; Roberts, C.L.; Dobbins, T.; Stavrou, E.; Black, K.; Morris, J.; Young, J. Incidence and outcomes of pregnancy-associated cancer in Australia, 1994–2008: A population-based linkage study. BJOG 2012, 119, 1572–1582.
- Bannister-Tyrrell, M.; Roberts, C.L.; Hasovits, C.; Nippita, T.; Ford, J.B. Incidence and outcomes of pregnancy-associated melanoma in New South Wales 1994–2008. Aust. N. Zeal. J. Obstet. Gynaecol. 2015, 55, 116–122.
- Lens, M.; Bataille, V. Melanoma in relation to reproductive and hormonal factors in women: Current review on controversial issues. Cancer Causes Control 2008, 19, 437–442.
- Donahue, T.; Lee, C.Y.; Sanghvi, A.; Obregon, R.; Sidiropoulos, M.; Cooper, C.; Merkel, E.A.; Yelamos, O.; Ferris, L.; Gerami, P. Immunosuppression is an independent prognostic factor associated with aggressive tumor behavior in cutaneous melanoma. J. Am. Acad. Dermatol. 2015, 73, 461–466.
- Fang, S.; Wang, Y.; Dang, Y.; Gagel, A.; Ross, M.I.; Gershenwald, J.E.; Cormier, J.N.; Wargo, J.; Haydu, L.E.; Davies, M.A.; et al. Association between body mass index, C-reactive protein levels, and Melanoma patient outcomes. J. Investig. Dermatol. 2017, 137, 1792–1795.
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322.
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body fatness and cancer--Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798.
- Secretan, B.; Straif, K.; Baan, R.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part E: Tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009, 10, 1033–1034.
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685.
- Le Dare, B.; Lagente, V.; Gicquel, T. Ethanol and its metabolites: Update on toxicity, benefits, and focus on immunomodulatory effects. Drug Metab. Rev. 2019, 51, 545–561.
- Xue, L.; Xu, F.; Meng, L.; Wei, S.; Wang, J.; Hao, P.; Bian, Y.; Zhang, Y.; Chen, Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS Lett. 2012, 586, 137–142.
- Mellor, D.D.; Hanna-Khalil, B.; Carson, R. A Review of the potential health benefits of low alcohol and alcohol-free beer: Effects of ingredients and craft brewing processes on potentially bioactive metabolites. Beverages 2020, 6, 25.
- Arranz, S.; Chiva-Blanch, G.; Valderas-Martinez, P.; Medina-Remon, A.; Lamuela-Raventos, R.M.; Estruch, R. Wine, beer, alcohol and polyphenols on cardiovascular disease and cancer. Nutrients 2012, 4, 759–781.
- Rivera, A.; Nan, H.; Li, T.; Qureshi, A.; Cho, E. Alcohol Intake and Risk of Incident Melanoma: A Pooled Analysis of Three Prospective Studies in the United States. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1550–1558.
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S. Regulation of cytochrome P450 2e1 expression by ethanol: Role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death Dis. 2013, 4, e554.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Alcohol consumption and ethyl carbamate. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 96, 3–1383.
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593.
- Cogliano, V.J.; Baan, R.; Straif, K.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; et al. Preventable exposures associated with human cancers. J. Natl. Cancer Inst. 2011, 103, 1827–1839.
- Koivisto, T.; Salaspuro, M. Acetaldehyde alters proliferation, differentiation and adhesion properties of human colon adenocarcinoma cell line Caco-2. Carcinogenesis 1998, 19, 2031–2036.
- Homann, N.; Karkkainen, P.; Koivisto, T.; Nosova, T.; Jokelainen, K.; Salaspuro, M. Effects of acetaldehyde on cell regeneration and differentiation of the upper gastrointestinal tract mucosa. J. Natl. Cancer Inst. 1997, 89, 1692–1697.
- Li, K.; Guo, W.; Li, Z.; Wang, Y.; Sun, B.; Xu, D.; Ling, J.; Song, H.; Liao, Y.; Wang, T.; et al. ALDH2 repression promotes lung tumor progression via accumulated acetaldehyde and DNA damage. Neoplasia 2019, 21, 602–614.
- Na, H.K.; Lee, J.Y. Molecular basis of alcohol-related gastric and colon cancer. Int. J. Mol. Sci. 2017, 18, 1116.
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943.
- Wang, Y.; Millonig, G.; Nair, J.; Patsenker, E.; Stickel, F.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology 2009, 50, 453–461.
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–193.
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox. Biol. 2014, 3, 56–62.
- Tsuruta, H.; Sonohara, Y.; Tohashi, K.; Aoki Shioi, N.; Iwai, S.; Kuraoka, I. Effects of acetaldehyde-induced DNA lesions on DNA metabolism. Genes Environ. 2020, 42, 2.
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell Longev. 2010, 3, 178–185.
- Abbas, O.; Mahalingam, M. Epidermal stem cells: Practical perspectives and potential uses. Br. J. Dermatol. 2009, 161, 228–236.
- Dohrman, D.P.; Diamond, I.; Gordon, A.S. Ethanol causes translocation of cAMP-dependent protein kinase catalytic subunit to the nucleus. Proc. Natl. Acad. Sci. USA 1996, 93, 10217–10221.
- Carlson, S.L.; Kumar, S.; Werner, D.F.; Comerford, C.E.; Morrow, A.L. Ethanol activation of protein kinase A regulates GABAA alpha1 receptor function and trafficking in cultured cerebral cortical neurons. J. Pharmacol. Exp. Ther. 2013, 345, 317–325.
- Kumar, S.; Ren, Q.; Beckley, J.H.; O’Buckley, T.K.; Gigante, E.D.; Santerre, J.L.; Werner, D.F.; Morrow, A.L. Ethanol activation of protein kinase A regulates GABA(A) receptor subunit expression in the cerebral cortex and contributes to ethanol-induced hypnosis. Front. Neurosci. 2012, 6, 44.
- Aroor, A.R.; Shukla, S.D. MAP kinase signaling in diverse effects of ethanol. Life Sci. 2004, 74, 2339–2364.
- Zeng, T.; Zhang, C.L.; Song, F.Y.; Zhao, X.L.; Yu, L.H.; Zhu, Z.P.; Xie, K.Q. PI3K/Akt pathway activation was involved in acute ethanol-induced fatty liver in mice. Toxicology 2012, 296, 56–66.
- Neasta, J.; Ben Hamida, S.; Yowell, Q.V.; Carnicella, S.; Ron, D. AKT signaling pathway in the nucleus accumbens mediates excessive alcohol drinking behaviors. Biol. Psychiatry 2011, 70, 575–582.
- Mercer, K.E.; Hennings, L.; Ronis, M.J. Alcohol consumption, Wnt/beta-catenin signaling, and hepatocarcinogenesis. Adv. Exp. Med. Biol. 2015, 815, 185–195.
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32.
- Wand, G.; Levine, M.; Zweifel, L.; Schwindinger, W.; Abel, T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J. Neurosci. 2001, 21, 5297–5303.
- Mantovani, G.; Bondioni, S.; Lania, A.G.; Rodolfo, M.; Peverelli, E.; Polentarutti, N.; Veliz Rodriguez, T.; Ferrero, S.; Bosari, S.; Beck-Peccoz, P.; et al. High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene 2008, 27, 1834–1843.
- Finger, E.C.; Castellini, L.; Rankin, E.B.; Vilalta, M.; Krieg, A.J.; Jiang, D.; Banh, A.; Zundel, W.; Powell, M.B.; Giaccia, A.J. Hypoxic induction of AKAP12 variant 2 shifts PKA-mediated protein phosphorylation to enhance migration and metastasis of melanoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4441–4446.
- Messing, R.O.; Petersen, P.J.; Henrich, C.J. Chronic ethanol exposure increases levels of protein kinase C delta and epsilon and protein kinase C-mediated phosphorylation in cultured neural cells. J. Biol. Chem. 1991, 266, 23428–23432.
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18.
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin. J. Cancer 2013, 32, 253–265.
- Xu, M.; Wang, S.; Qi, Y.; Chen, L.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Shi, X.; Luo, J. Role of MCP-1 in alcohol-induced aggressiveness of colorectal cancer cells. Mol. Carcinog. 2016, 55, 1002–1011.
- Hong, Z.; Fang, Z.; Lei, J.; Shi, G.; Zhang, Y.; He, Z.; Li, B.W.; Zhong, S. The significance of Runx2 mediating alcohol-induced Brf1 expression and RNA Pol III gene transcription. Chem. Biol. Interact. 2020, 323, 109057.
- Yi, Y.; Huang, C.; Zhang, Y.; Tian, S.; Lei, J.; Chen, S.; Shi, G.; Wu, Z.; Xia, N.; Zhong, S. Exploring a common mechanism of alcohol-induced deregulation of RNA Pol III genes in liver and breast cells. Gene 2017, 626, 309–318.
- Barron, K.A.; Jeffries, K.A.; Krupenko, N.I. Sphingolipids and the link between alcohol and cancer. Chem. Biol. Interact. 2020, 322, 109058.
- Serio, R.N.; Gudas, L.J. Modification of stem cell states by alcohol and acetaldehyde. Chem. Biol. Interact. 2020, 316, 108919.
- Di Rocco, G.; Baldari, S.; Pani, G.; Toietta, G. Stem cells under the influence of alcohol: Effects of ethanol consumption on stem/progenitor cells. Cell Mol. Life Sci. 2019, 76, 231–244.
- Subramaniyan, V.; Chakravarthi, S.; Jegasothy, R.; Seng, W.Y.; Fuloria, N.K.; Fuloria, S.; Hazarika, I.; Das, A. Alcohol-associated liver disease: A review on its pathophysiology, diagnosis and drug therapy. Toxicol. Rep. 2021, 8, 376–385.
- Bode, C.; Bode, J.C. Alcohol’s role in gastrointestinal tract disorders. Alcohol Health Res. World 1997, 21, 76–83.
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009, 6, e50.
- Slutske, W.S.; Heath, A.C.; Madden, P.A.; Bucholz, K.K.; Dinwiddie, S.H.; Dunne, M.P.; Statham, D.S.; Whitfield, J.B.; Martin, N.G. Is alcohol-related flushing a protective factor for alcoholism in Caucasians? Alcohol Clin. Exp. Res. 1995, 19, 582–592.
- Gill, K.; Eagle Elk, M.; Liu, Y.; Deitrich, R.A. An examination of ALDH2 genotypes, alcohol metabolism and the flushing response in Native Americans. J. Stud. Alcohol 1999, 60, 149–158.
- Higgins, E.; du Vivier, A. Alcohol intake and other skin disorders. Clin. Dermatol. 1999, 17, 437–441.
- Liu, S.W.; Lien, M.H.; Fenske, N.A. The effects of alcohol and drug abuse on the skin. Clin. Dermatol. 2010, 28, 391–399.
- Nyman, E.; Palmlöv, A. The elimination of ethyl alcohol in sweat 1. Skand. Arch. Für Physiol. 1936, 74, 155–159.
- Pawan, G.L.; Grice, K. Distribution of alcohol in urine and sweat after drinking. Lancet 1968, 2, 1016.
- Baker, L.B.; Wolfe, A.S. Physiological mechanisms determining eccrine sweat composition. Eur. J. Appl. Physiol. 2020, 120, 719–752.
- Kwak, S.; Brief, E.; Langlais, D.; Kitson, N.; Lafleur, M.; Thewalt, J. Ethanol perturbs lipid organization in models of stratum corneum membranes: An investigation combining differential scanning calorimetry, infrared and (2)H NMR spectroscopy. Biochim. Biophys. Acta 2012, 1818, 1410–1419.
- Hook-Nikanne, J.; Kariniemi, A.L.; Renkonen, O.V.; Mustakallio, K.; Salaspuro, M. Could bacterial acetaldehyde production explain the deleterious effect of alcohol on skin diseases? Acta Derm. Venereol. 1995, 75, 330.
- Destek, S.; Gul, V.O.; Ahioglu, S.; Erbil, Y. A rare disease of the digestive tract: Esophageal Melanosis. Gastroenterol. Res. 2016, 9, 56–60.
- Siddiqi, A.; Chaudhary, F.S.; Naqvi, H.A.; Saleh, N.; Farooqi, R.; Yousaf, M.N. Black esophagus: A syndrome of acute esophageal necrosis associated with active alcohol drinking. BMJ Open Gastroenterol. 2020, 7, e000466.
- Nagra, N.; Tolentino, L.; Singhvi, G. Esophageal Melanosis: A rare condition of undetermined significance. Clin. Gastroenterol. Hepatol. 2020, 18, e59.
- Jin, S.; Chen, J.; Chen, L.; Histen, G.; Lin, Z.; Gross, S.; Hixon, J.; Chen, Y.; Kung, C.; Chen, Y.; et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 9088–9093.
- Matsumoto, A.; Ito, S.; Wakamatsu, K.; Ichiba, M.; Vasiliou, V.; Akao, C.; Song, B.J.; Fujita, M. Ethanol induces skin hyperpigmentation in mice with aldehyde dehydrogenase 2 deficiency. Chem. Biol. Interact. 2019, 302, 61–66.
- Matsumura, Y.; Li, N.; Alwaseem, H.; Pagovich, O.E.; Crystal, R.G.; Greenblatt, M.B.; Stiles, K.M. Systemic adeno-associated virus-mediated gene therapy prevents the multiorgan disorders associated with Aldehyde Dehydrogenase 2 deficiency and chronic ethanol ingestion. Hum. Gene Ther. 2020, 31, 163–182.
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923.
- Appenzeller, B.M.; Agirman, R.; Neuberg, P.; Yegles, M.; Wennig, R. Segmental determination of ethyl glucuronide in hair: A pilot study. Forensic Sci. Int. 2007, 173, 87–92.
- D’Alessandro, A.; Fu, X.; Reisz, J.A.; Stone, M.; Kleinman, S.; Zimring, J.C.; Busch, M.; for the Recipient Epidemiology and Donor Evaluation Study-III (REDS III). Ethyl glucuronide, a marker of alcohol consumption, correlates with metabolic markers of oxidant stress but not with hemolysis in stored red blood cells from healthy blood donors. Transfusion 2020, 60, 1183–1196.
- Lewis, S.S.; Hutchinson, M.R.; Zhang, Y.; Hund, D.K.; Maier, S.F.; Rice, K.C.; Watkins, L.R. Glucuronic acid and the ethanol metabolite ethyl-glucuronide cause toll-like receptor 4 activation and enhanced pain. Brain Behav. Immun. 2013, 30, 24–32.
- Mandrekar, P.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Szabo, G. Inhibition of myeloid dendritic cell accessory cell function and induction of T cell anergy by alcohol correlates with decreased IL-12 production. J. Immunol. 2004, 173, 3398–3407.
- Ness, K.J.; Fan, J.; Wilke, W.W.; Coleman, R.A.; Cook, R.T.; Schlueter, A.J. Chronic ethanol consumption decreases murine Langerhans cell numbers and delays migration of Langerhans cells as well as dermal dendritic cells. Alcohol Clin. Exp. Res. 2008, 32, 657–668.
- Parlet, C.P.; Schlueter, A.J. Mechanisms by which chronic ethanol feeding impairs the migratory capacity of cutaneous dendritic cells. Alcohol Clin. Exp. Res. 2013, 37, 2098–2107.
- Lecomte, E.; Grolier, P.; Herbeth, B.; Pirollet, P.; Musse, N.; Paille, F.; Braesco, V.; Siest, G.; Artur, Y. The relation of alcohol consumption to serum carotenoid and retinol levels. Effects of withdrawal. Int. J. Vitam. Nutr. Res. 1994, 64, 170–175.
- Stahl, W.; Sies, H. Carotenoids and protection against solar UV radiation. Skin Pharmacol. Appl. Skin Physiol. 2002, 15, 291–296.
- Saladi, R.N.; Nektalova, T.; Fox, J.L. Induction of skin carcinogenicity by alcohol and ultraviolet light. Clin. Exp. Dermatol. 2010, 35, 7–11.
- Darvin, M.E.; Sterry, W.; Lademann, J.; Patzelt, A. Alcohol consumption decreases the protection efficiency of the antioxidant network and increases the risk of sunburn in human skin. Skin Pharmacol. Physiol. 2013, 26, 45–51.
- Darvin, M.E.; Patzelt, A.; Knorr, F.; Blume-Peytavi, U.; Sterry, W.; Lademann, J. One-year study on the variation of carotenoid antioxidant substances in living human skin: Influence of dietary supplementation and stress factors. J. Biomed. Opt. 2008, 13, 044028.
- Leo, M.A.; Lieber, C.S. Alcohol, vitamin A, and beta-carotene: Adverse interactions, including hepatotoxicity and carcinogenicity. Am. J. Clin. Nutr. 1999, 69, 1071–1085.
- Millen, A.E.; Tucker, M.A.; Hartge, P.; Halpern, A.; Elder, D.E.; Guerry, D.t.; Holly, E.A.; Sagebiel, R.W.; Potischman, N. Diet and melanoma in a case-control study. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1042–1051.
- Brand, R.M.; Stottlemyer, J.M.; Paglia, M.C.; Carey, C.D.; Falo, L.D., Jr. Ethanol consumption synergistically increases ultraviolet radiation induced skin damage and immune dysfunction. J. Dermatol. Sci. 2021, 101, 40–48.
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304.
- Ji, C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem. Res. Int. 2012, 2012, 216450.
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784.
- Strickland, F.M.; Muller, H.K.; Stephens, L.C.; Bucana, C.D.; Donawho, C.K.; Sun, Y.; Pelley, R.P. Induction of primary cutaneous melanomas in C3H mice by combined treatment with ultraviolet radiation, ethanol and aloe emodin. Photochem. Photobiol. 2000, 72, 407–414.
- Strickland, F.M.; Pathak, S.; Multani, A.S.; Pelley, R.P.; Donawho, C.K. Molecular characterization of new melanoma cell lines from C3H mice induced by ethanol plus ultraviolet radiation. Cancer Res. 2003, 63, 3503–3510.
- Wikonkal, N.M.; Brash, D.E. Ultraviolet radiation induced signature mutations in photocarcinogenesis. J. Investig. Dermatol. Symp. Proc. 1999, 4, 6–10.
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499.
- Vu, H.L.; Aplin, A.E. Targeting mutant NRAS signaling pathways in melanoma. Pharmacol. Res. 2016, 107, 111–116.
- Fargnoli, M.C.; Chimenti, S.; Keller, G.; Soyer, H.P.; Dal Pozzo, V.; Hofler, H.; Peris, K. CDKN2a/p16INK4a mutations and lack of p19ARF involvement in familial melanoma kindreds. J. Investig. Dermatol. 1998, 111, 1202–1206.
- Meadows, G.G.; Zhang, H. Effects of alcohol on tumor growth, metastasis, immune response, and host survival. Alcohol Res. 2015, 37, 311–322.
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell Mol. Life Sci. 2015, 72, 1249–1260.
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318.
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122.
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98.
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 1998, 391, 298–301.
- Hauge, C.; Frodin, M. RSK and MSK in MAP kinase signalling. J. Cell Sci. 2006, 119, 3021–3023.
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637.
- Kashyap, T.; Pramanik, K.K.; Nath, N.; Mishra, P.; Singh, A.K.; Nagini, S.; Rana, A.; Mishra, R. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3beta signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. 2018, 86, 234–243.
- Kim, J.H.; Hong, A.R.; Kim, Y.H.; Yoo, H.; Kang, S.W.; Chang, S.E.; Song, Y. JNK suppresses melanogenesis by interfering with CREB-regulated transcription coactivator 3-dependent MITF expression. Theranostics 2020, 10, 4017–4029.
- Tan, W.; Bailey, A.P.; Shparago, M.; Busby, B.; Covington, J.; Johnson, J.W.; Young, E.; Gu, J.W. Chronic alcohol consumption stimulates VEGF expression, tumor angiogenesis and progression of melanoma in mice. Cancer Biol. Ther. 2007, 6, 1211–1217.
- Blank, S.E.; Meadows, G.G. Ethanol modulates metastatic potential of B16BL6 melanoma and host responses. Alcohol Clin. Exp. Res. 1996, 20, 624–628.
- Kushiro, K.; Nunez, N.P. Ethanol inhibits B16-BL6 melanoma metastasis and cell phenotypes associated with metastasis. Vivo 2012, 26, 47–58.
- Zhang, H.; Zhu, Z.; McKinley, J.M.; Meadows, G.G. IFN-gamma is essential for the inhibition of B16BL6 melanoma lung metastasis in chronic alcohol drinking mice. Clin. Exp. Metastasis 2011, 28, 301–307.
- Meadows, G.G.; Elstad, C.A.; Blank, S.E.; Gallucci, R.M.; Pfister, L.J. Alcohol consumption suppresses metastasis of B16-BL6 melanoma in mice. Clin. Exp. Metastasis 1993, 11, 191–199.
- Abdallah, M.A.; Singal, A.K. Mitochondrial dysfunction and alcohol-associated liver disease: A novel pathway and therapeutic target. Signal Transduct. Target Ther. 2020, 5, 26.
- Bonet-Ponce, L.; Saez-Atienzar, S.; da Casa, C.; Flores-Bellver, M.; Barcia, J.M.; Sancho-Pelluz, J.; Romero, F.J.; Jordan, J.; Galindo, M.F. On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. Biochim. Biophys. Acta 2015, 1852, 1400–1409.
- Dal Yontem, F.; Kim, S.H.; Ding, Z.; Grimm, E.; Ekmekcioglu, S.; Akcakaya, H. Mitochondrial dynamic alterations regulate melanoma cell progression. J. Cell Biochem. 2018, 120, 2098–2108.
- From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO); Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int. J. Stroke 2018, 13, 612–632.
- Sadikot, R.T.; Bedi, B.; Li, J.; Yeligar, S.M. Alcohol-induced mitochondrial DNA damage promotes injurious crosstalk between alveolar epithelial cells and alveolar macrophages. Alcohol 2019, 80, 65–72.
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488.
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830.
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118.
- Molina, P.E.; Happel, K.I.; Zhang, P.; Kolls, J.K.; Nelson, S. Focus on: Alcohol and the immune system. Alcohol Res. Health 2010, 33, 97–108.
- Zhang, H.; Zhu, Z.; Meadows, G.G. Chronic alcohol consumption impairs distribution and compromises circulation of B cells in B16BL6 melanoma-bearing mice. J. Immunol. 2012, 189, 1340–1348.
- Zhang, H.; Zhu, Z.; Meadows, G.G. Chronic alcohol consumption decreases the percentage and number of NK cells in the peripheral lymph nodes and exacerbates B16BL6 melanoma metastasis into the draining lymph nodes. Cell Immunol. 2011, 266, 172–179.
- Zhang, H.; Meadows, G.G. Chronic alcohol consumption enhances myeloid-derived suppressor cells in B16BL6 melanoma-bearing mice. Cancer Immunol. Immunother. 2010, 59, 1151–1159.
- Rappa, G.; Anzanello, F.; Lorico, A. Ethanol induces upregulation of the nerve growth factor receptor CD271 in human melanoma cells via nuclear factor-kappaB activation. Oncol. Lett. 2015, 10, 815–821.
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109.
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Publisher Correction: The low affinity neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat. Commun. 2018, 9, 314.
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459.
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased CD271 expression by the NF-kB pathway promotes melanoma cell survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015, 1, 15030.
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in Melanoma. Cell 2018, 174, 843–855.e19.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435.
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978.
- Boshuizen, J.; Vredevoogd, D.W.; Krijgsman, O.; Ligtenberg, M.A.; Blankenstein, S.; de Bruijn, B.; Frederick, D.T.; Kenski, J.C.N.; Parren, M.; Bruggemann, M.; et al. Reversal of pre-existing NGFR-driven tumor and immune therapy resistance. Nat. Commun. 2020, 11, 3946.
- Furuta, J.; Inozume, T.; Harada, K.; Shimada, S. CD271 on melanoma cell is an IFN-gamma-inducible immunosuppressive factor that mediates downregulation of melanoma antigens. J. Investig. Dermatol. 2014, 134, 1369–1377.
- Mehta, A.; Kim, Y.J.; Robert, L.; Tsoi, J.; Comin-Anduix, B.; Berent-Maoz, B.; Cochran, A.J.; Economou, J.S.; Tumeh, P.C.; Puig-Saus, C.; et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. 2018, 8, 935–943.
More