CThe endocannancer is a disease which affects approximately 40% of people in their lifetime. Chemotherapy, the primary choice for treatment of cancer, is often ineffective or/and presents itself with many debilitating side effects, including loss of appetite, nausea, insomnia, and anxiety. Cbinoid system (ECS) is an ancient homeostasis mechanism operating from embryonic stages to adulthood. It controls the growth and development of many cells and cell lineages. Dysregulation of the components of cannabis extracts, including cannabinoids and terpenes, may present an alternative for the ECS may result in uncontrolling side effects and may be used for tumor shrinkage together with chemodrugs. Cannabinoids act on so called endocannabinoid system (ECS) that operates in human body to maintain homeostasis. ECS promotes healthyed proliferation, adhesion, invasion, inhibition of apoptosis and increased vascularization, leading to the development of tissues and regulates many processes in our organism and when disbalanced may lead to disease, including cancervarious malignancies. Cancer is the disease of uncontrolled cell division.
- endocannabinoid system
- cancer and carcinogenesis
- primary care
- palliative care
- cannabinol
- tetrahydrocannabinol
1. Introduction
2. Role of Endocannabinoids in the Human Body
ECS is active in virtually all cells of houmanr organism. It plays an important role in the reproduction, function, and proper development of gametes [7], fertilization event, embryo implantation, and proper placenta development [8]. It is also active at all stages of embryogenesis, regulating cell division, and tissue and organ development, specifically, regulating differentiation of neural progenitors, synaptogenesis, and axonal migration [9]. During human adult life, it regulates homeostasis of many tissues, playing critical role in proper brain function by regulating neuronal synaptic communications affecting critical organismal functions, including general metabolism, growth and development, reproduction, learning and memory formation, mood, and behavior, among others [10]. In the peripheral tissues, endocannabinoids are involved in endocrine regulation and energy balance [11], as well as regulating the function of innate and adaptive immune system and immune response [12], regulating cell migration and apoptosis. The activity and functionality of ECS depends on many factors, from cell- and tissue-specific differences in the synthesis of endocannabinoids, to the number and the activity of endocannabinoid and auxiliary receptors, to the expression and the activity of enzymes involved in the degradation of circulating endocannabinoids. In the cells, endocannabinoids acting in CB-receptor-dependent and independent manner exhibit anti-oxidative properties, are involved in clearance of damaged molecules and regulate mitochondrial activity. Anti-oxidative properties are associated with the inhibition of production of reactive oxygen species (ROS), metal chelation and prevention/alleviation of ROS-induced cell damage [13]. It should be noted that the anti-oxidative effects of cannabinoids are cell specific—while in most cells of the body, they mitigate oxidative stress, in hepatic cells they may cause it, leading to cell death [14]. Similarly, in cancer cells, such as gliomas and leukemia, cannabinoids promote oxidative stress [13]. Cannabinoids contribute to recycling of damaged molecules and are likely involved in autophagy in health tissues [15]—the activity well documented in cancer cells (discussed below). In normal cells, they increase lysosomal stability and integrity [15] through CB1 receptors found on the surface of lysosomes. CB1 receptors are also present on the surface of mitochondria. They regulate mitochondrial oxidative phosphorylation in a positive and a negative manner, acting through the CB1 receptor, but it is not clear what modulates this activity [13]. When cells are stressed, cannabinoids attenuate mitochondrial damage [16] and decrease calcium-induced cytochrome c release [17].2.1. Mechanism of Action—Ligand/Receptor
Cannabinoid receptors are ubiquitous and expressed on the cell surface as well on cell organelles, including mitochondria and lysosomes. Classical cannabinoid receptors include CB1 and CB2. CB1 is expressed at a higher level in central and peripheral nervous systems, while CB2 is expressed in many different tissues, including the immune system, internal organs, skin, bone, muscle, and glia in the brain [18]. CB1 and CB2 are GPCR (Gi/o) protein-coupled receptors, and when activated, they modulate various cellular functions through receptor internalization; interaction with other G-protein-coupled receptors; inhibition of adenylyl cyclase activity, changing the activity of calcium and potassium channels; increasing phosphorylation of various mitogen-activated protein kinases (MAPK); and many more functions [12] (Figure 1).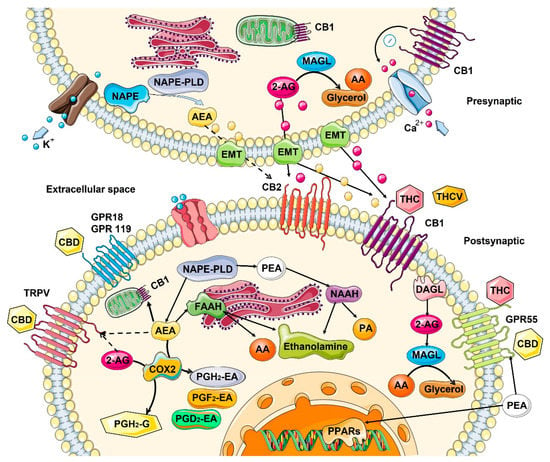
2.2. Role in the Control of Cell Division and Cell Proliferation
It appears that ECS controls the fate of many cells in the organism, regulating the cell division and proliferation, apoptosis, necrosis and autophagy in several organs and organ systems, including the brain, skin, and immune system. In the central nervous system (CNS), the ECS system functions as a neuroprotective system that controls glutamate excitotoxicity, calcium influx, inflammation, and autophagy [25]. In the CNS, the interaction of endocannabinoids with CB1/CB2 and other receptors mediates synaptic plasticity or progenitor cell fate in the central nervous system, promoting self-repair of the brain [26]. It also appears that constitutive release of 2-arachidonoylglycero by late oligodendrocyte progenitors allows oligodendrocyte maturation by activating CB receptors and downstream ERK pathway [27]. In skin, ECS activity maintains the cutaneous homeostasis through the regulation of skin cell proliferation, survival, and differentiation [28]. Locally produced AEA inhibits the cellular growth and the differentiation of cultured NHEK and HaCaT keratinocytes, as well as inducing apoptosis of human HaCaT keratinocytes [28]. CB1 activity is higher in differentiated skin layers [29]. In human cultured hair follicles, AEA but not 2-AG inhibit elongation and proliferation of hair shaft and induce intraepithelial apoptosis in a CB1-dependent manner [30]. Both AEA and 2-AG induce apoptosis of human sebaceous-gland-derived SZ95 sebocytes in a CB2-dependent manner [31]. In the immune system, the central role is played by CB2 receptors that are mainly expressed by cells (T and B lymphocytes) and peripheral tissues of the immune system (spleen and thymus) where it regulates immune suppression, apoptosis, and cell migration [32]. In in vitro studies, it was demonstrated that anandamide inhibits mitogen-induced proliferation of T cells [33], while inhibiting the chemokine SDF-1-induced migration of CD8+ T cells [34]. In contrast, 2-AG, but not anandamide, induced CB2-dependent migration in natural killer cell line KHYG-1 cells [35]. In B cells, 2-AG chemo-attracts naïve B cells and marginal zone B cells and inhibits the function of activated B cells, while 2-AG and anandamide suppress the migration of neutrophils [36]. Additionally, anandamide induces the apoptosis of murine bone-marrow-derived DCs (BMDCs) in a CB1- and CB2-dependent manner [37].2.3. Changes in the ECS with Age
Cancer can be considered an age-associated disease, due to the accumulation of cellular and DNA damage. From this perspective, it is interesting to understand what happens to ECS with age. In general, information about age-related changes in the ECS is scarce. Most of the data are related to changes in the central nervous system, and even then, the data are very contradictory. In general, it is believed that the activity of ECS declines with age [13]. In rats, in one study, a general decrease in the expression of CB1 and a decrease in density of the receptors in various brain areas with age was observed [38], while in another study—in which only redistribution of the receptors was noted– they were reduced in the postrhinal, but elevated in the entorhinal and temporal cortices in old animals [39] (Table 1). In mice, no changes in the receptor density in most brain regions was found with age, but instead, a significantly reduced receptor/Gi protein coupling was observed [40]. In one study on humans, CB1 expression increased, predominantly in females, most drastically in the basal ganglia, the lateral temporal cortex, and in the hippocampus [41], while another study reported no change [39]. As for endocannabinoids, the picture is not clear either—some studies suggested a decrease, while others found no difference in different brain regions of young and old animals. [13]. However, animals lacking FAAH—the enzyme degrading anandamide showed less pronounced features of aging—decreased expression of pro-inflammatory genes and decreased decline in cardiac function [42] (Table 1).| Tissues/Organs | Endocannabinoids | Receptors | Metabolizing Enzymes |
|---|---|---|---|
| Skin | No reliable data | ↓ in CB1 expression [13] | FAAH tends to ↓ with age [43][45] |
| Lung | 2-AG ↓ and AEA ↑ in mice [44][46] | No reliable data | No reliable data |
| Brain | From no change [40] to a ↓ in AEA [45][47] ↓ in 2-AG levels in mice [46][48] |
From ↑ in humans [41] to no change [39] to a ↓ [38][47][38,49] in mice/rats in CB1 expression, brain area-specific | ↓ FAAH activity in rats [48][50] ↑ in MAGL levels in mice [46][48] |
| Blood | Small ↑ in 2-AG and AEA in mice [44][46] | No reliable data | No reliable data |
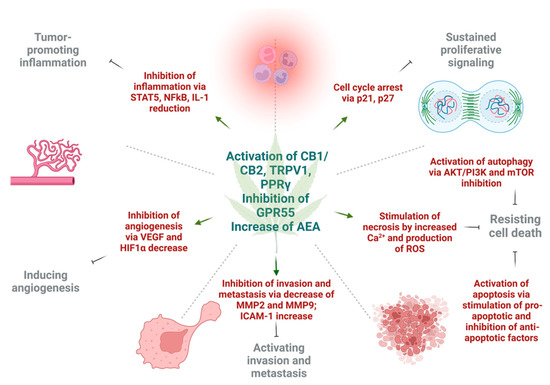
3. Effect of Cannabinoids on Various Hallmarks of Cancer
Various in vitro and in vivo experiments have shown that cannabinoids can target almost every hallmark of cancer (Figure 2) [51][186]. They inhibit proliferation, reduce inflammation, stimulate apoptosis, and inhibit tumor invasiveness, angiogenesis, and metastasis [52][53][54][55][187,188,189,190]. One of the most important effects of cannabinoids, besides their antitumor ability, is that they are less likely to affect non-transformed normal cells surrounding tumors, and they may even have protective effects. For instance, cannabinoids may induce cell death in glioma cells while protecting normal astroglial and oligodendroglial cells from apoptosis via CB1 receptors [52][187]. Studies on animals show the protective effects of cannabinoids against certain types of tumors. For example, a dose-dependent decrease in the incidence of hepatic adenomas and hepatocellular carcinomas in mice that were given THC over 2 years was noted. Additionally, lower incidence rates of benign tumors in mammary glands, uterus, testis, and pancreas were seen in tested rats [56][191].